
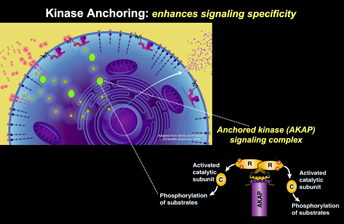
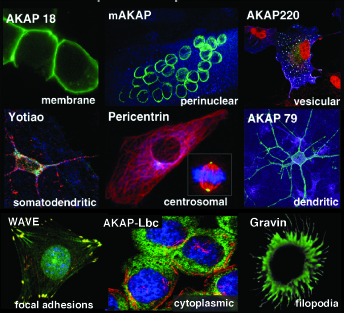
The AKAP Model
Generation of the second-messenger cAMP following receptor activation leads to activation of Protein Kinase A (PKA) and the subsequent phosphorylation of a variety of cellular substrates. Where and when PKA becomes active determines the specificity of a given response. We have shown that the subcellular targeting of PKA occurs through association with an A-Kinase Anchoring Protein (AKAP). AKAPs are a functionally related family of more than 50 distinct proteins defined by their ability to bind to the PKA holoenzyme. Each AKAP contains at least two functional motifs: anchoring PKA and subcellular targeting. The first is a conserved amphipathic helix that slots into a hydrophobic pocket formed by the amino terminus of the PKA regulatory subunit (RII) dimer. Each AKAP also contains a unique targeting domain that directs the kinase-AKAP complex to distinct intracellular sites. Recently we have explored the ramifications of structural constraints of anchored PKA action.
Classical in vitro biochemistry studies of this kinase led to the textbook view that active PKA catalytic (C) subunits dissociate from the regulatory (R) subunits in the presence of cAMP. This creates a paradox because the subcellular targeting function of AKAPs would be lost if the C subunit is free to diffuse away. We employed structural biology approaches to resolve this paradox. Single particle negative stain EM studies elucidated the topology of an AKAP18-PKA holoenzyme (RIIdimer and two C subunits) complex. This work redefined the parameters of anchored PKA action as it identified the complex as clusters of three densities in a range of configurations, from tightly-packed symmetric triangular arrays to fully-extended linear configurations. The inherent flexibility within this complex sets a dynamic range of distance for basal and cAMP-dependent phosphorylation. Movement of anchored C subunit augments phosphorylation of local substrates either interacting with the AKAP or near the peripheral lobes of the extended holoenzyme. An unexpected implication of this revised structural model challenges prevailing dogma: even though cAMP production stimulates kinase activity, AKAP-PKA holoenzymes do not dissociate and remain functionally intact inside cells. This ‘signaling island’ concept radically changes our view of how AKAP complexes operate and indicates that protein phosphorylation is much more regionally confined than previously appreciated.
AKAP Signaling Networks
Many AKAPs are also multivalent scaffold proteins that associate with numerous signaling enzymes including other kinases, protein phosphatases, phosphodiesterases and substrates to form signaling complexes. Distinct combinations of anchored enzymes allow these complexes to respond to a variety of second messenger–mediated signals and enable convergence of signaling pathways in a context specific manner.
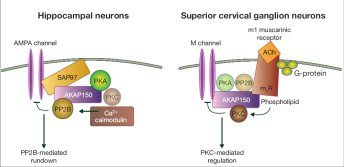
Several years ago we demonstrated that AKAPs simultaneously bind enzymes with opposing actions, such as kinases and phosphatases to target entire signaling complexes to specific substrates. One prototypic example is the neuronal anchoring protein, AKAP79, that targets PKA, protein kinase C (PKC), and the calcium/calmodulin-dependent phosphatase (PP2B) to sites in the membrane. Thus, AKAP79 provides a point of convergence for multiple second messenger signals, such as cAMP, Ca++, and phospholipids. Furthermore, AKAP79 can assemble distinct macromolecular complexes within different cellular contexts. In hippocampal neurons AKAP79 forms a complex with AMPA receptors in which PKA phosphorylation and Ca++/calmodulin regulated PP2B dephosphorylation work together to regulate channel activity. However, in superior cervical ganglion neurons the same AKAP interacts with M-channels and it is the AKAP79-bound PKC that is the binding partner that regulates suppression of the currents. Furthermore, our work has shown that interaction with AKAP79 influences how binding partners such as PKC respond to certain pharmacological inhibitors. AKAP79 protects this kinase from inhibition with certain ATP competitive inhibitors. Therefore, intracellular binding partners not only couple individual molecular events in a cell signaling process, but can also change the pharmacological profile of an enzyme.
Consequently, AKAPs not only control access to substrates, but may also influence the response of anchored enzymes to certain small-molecule inhibitors. We focused on the synchronization of phosphorylation events at the centrosome during mitosis following our discovery that an AKAP known as Gravin integrates mitogenic and second messenger kinases at the mitotic spindle to drive this dynamic stage of the cell cycle. Mass spectrometry, molecular and cellular approaches establish that the kinase CDK1/Cyclin B1 phosphorylates Gravin at threonine-766 to prime the recruitment of polo-like kinase, Plk1, at defined phases of mitosis. Furthermore, tumor sections from glioblastoma patients revealed increased levels of phospho-T766 Gravin and robust recruitment of active Plk1 to the mitotic spindle. Loss of control of this transient kinase-anchoring event has important pathological ramifications as Gravin transcripts are also reduced in prostate and ovarian cancers. Functional studies confirmed that manipulating elements of this Gravin-kinase scaffold adversely impacts astral microtubule assembly, promotes spindle misorientation and delays mitosis. Following up on this new area of study we will explore whether targeting of inhibitor drugs to organelles can provide a new pharmacological strategy borne of our improved understanding of AKAP nano-compartments.
AKAP's coordinate complex biological processes
Initially discovered as simple binding proteins purported to direct PKA to specific locations, we now appreciate that AKAPs comprise a large family of multivalent enzyme scaffolds that synchronize many complex cellular events. They constrain broad-specificity enzymes in customized macromolecular signaling islands to enable cells to respond efficiently and accurately to the ebb and flow of diffusible second-messenger signals. Early studies demonstrated that anchored PKA plays a role in the regulation of cardiac and skeletal muscle L-type Ca++ channels and neuronal N-methyl-D-aspartate (NMDA) receptor channels. We have gone on to discover that AKAPs also regulate PKA function in more complicated physiological systems, such as insulin secretion from pancreatic islet β cells and the role of kinase and phosphatase anchoring in insulin homeostasis.
An illustrative example of this latter concept is provided by recent work on AKAP-mediated control of comorbidities associated with diabetes and hypertension in our aging population. This led us to investigate the role of AKAPs in systemic control of water homeostasis and renal pathologies. Reabsorption of water from the luminal fluid of the nephron occurs through aquaporin-2 (AQP2) water pores in principal cells that line the kidney-collecting duct. Bi-directional control of AQP2 trafficking to apical membranes of these cells is responsive to the hormone vasopressin, anchored PKA and targeted phosphatases. Formation of an “actin barrier” that impedes transit of AQP2 to the apical membrane is vital to this process. We recently identified that AKAP220 coordinates a macro-molecular complex of signaling enzymes and effector proteins that regulate actin polymerization near the cell cortex, direct AQP2 localization and promote water excretion. These discoveries provide a link to kidney physiology when considered along with our evidence that AKAP220 also interacts with AQP2 water pores. CRISPR/Cas9 gene-editing and super resolution microscopy show that loss of AKAP220 disrupts apical actin networks in 3D organoid cultures from kidney-derived IMCD3 cells. Similar defects are evident in kidney tissue sections from mice that do not have the AKAP220 gene. Measurement of urine osmolality revealed that these mice fail to respond effectively to overhydration. This systemic defect in water homeostasis is likely related to altered trafficking of AQP2 within these cells. SIM imaging of kidney sections from AKAP220-deficient mice provided support for this model, revealing that RhoA, a GTPase modulator of the actin cytoskeleton and AQP2 accumulate at the apical surface of the kidney-collecting duct. Thus AKAP220 constrains membrane-proximal signaling complexes to impact actin barrier dynamics and AQP2 trafficking in kidney collecting ducts.