
Bioengineering Department, Box 355061, University of Washington, Seattle, WA 98195, USA

|
|
Bioengineering Department, Box 355061, University of Washington, Seattle, WA 98195, USA |
|
Delivering therapeutic drugs to the intended site of action at a controlled rate is an important goal of modern medicine. It is now well understood that controlling the rate of release of drugs can have several positive effects on the long-term outcome for the patient. Recent work has centered on fabrication of macroscopic devices that allow continuous release of some drugs under limited circumstances. We have begun a novel approach to continuous release in which neither macroscopic matrix nor pump is required, and the "device fabrication" is accomplished through molecular-level self-assembly. The long-term objective of the work is to develop a device-free method by which many drugs can be released into the body in a continuous manner (0th-order kinetics) through association with self-assembling tubular microstructures.
Lipid tubules are a recently discovered self-organizing system in which lipids crystallize into tightly packed bilayers that spontaneously form hollow cylinders less than 1 µm in diameter. Because of the tight packing of the surfactants in tubules, the microstructures should dissolve (or be enzymatically degraded) from their ends only. Since the size of the end (the only available surface area for removal of drug) is constant until the tubule is annihilated, a population of tubules of uniform length will release surfactant at a constant rate. Since the wide range of two-chain surfactants now known to form tubules includes lipidated polypeptides, and such lipidated polypeptides are composed of innocuous nontoxic biomolecules, it should be possible to synthesize tubule-forming lipids with polar headgroups that contain polypeptide and non-polypeptide drugs. These lipidated drugs might be active themselves if the hydrophobic groups do not interfere with binding at their sites of action; alternatively, the tubule-forming drugs might function as prodrugs that become active when their hydrophobic moieties are cleaved. The aforementioned tight packing of the lipids will be transferred to the drug headgroups. Such structures will either dissolve spontaneously at a rate controlled by structure-determined solubility of the lipidated prodrug molecules, or they might release drug by enzymatic degradation of the microstructures from their ends. These "prodrug tubules" could function as continuous release systems independent of any macroscopic encapsulation or delivery devices.
Tubule-based continuous release would probably have first applications in topical administration of drugs, antibacterials and antifungals, release of antitumor drugs/peptides by injections of tubules into or near tumors, antibiotic and growth factor delivery for wound dressings, long-acting vaccines, delivery of peptide and non-peptide drugs from subcutaneous sites, and in vitro use for tissue/cell culture factors.
The short-term objective of this project is to perform in vitro experiments that prove or disprove the following 3 hypotheses:
A homogeneous population of lipid tubules will dissolve (or be enzymatically degraded) in such a manner that the rate of release of the constituent molecules (or parts thereof) will be constant until the microstructures are consumed-
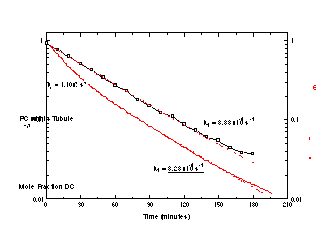
We have demonstrated this for the case of enzymatic hydrolysis of diacetylenic phospholipid tubules, and have filed a patent on the basis of this demonstration. The PLA2 hydrolysis products are, however, not water soluble, and we have found circumstances in which hydrolysis is not strictly 0th order. Our one case of study of dissolution in the presence of detergents shows 1st order kinetics. Work has just begun on monitoring the detergent-free dissolution of tubules and helices.
Ligation to an appropriate hydrophobic anchoring moiety will allow both model water-soluble molecules and clinically significant drugs and prodrugs to self-associate into tubular microstructures. Of particular interest are bioactive polypeptides.
We have synthesized peptide-based surfactants that include trypsin-sensitive cleavage sites in their headgroups, and have shown that they can form tubules and helices. Enzymatic cleavage studies are just beginning.
Tubule-associated drugs or prodrugs will continuously release drugs in simulations of drug delivery environments, either through dissolution of the molecules from tubule ends or through enzymatic cleavage from the tubules.
Preliminary studies of tubule stability in vivo and in in vitro simulants of extracellular fluids have been performed. All materials and methods are in position to begin studies of the release of drug simulants.
The Gelb and Yager groups provide complementary expertise in organic synthesis, enzymatic degradation of lipid bilayers and the self-assembly of lipids into tubules. Primary techniques employed are organic synthesis, differential scanning calorimetry, optical and electron microscopies, Raman spectroscopy, and enzyme and dissolution kinetic analyses. The work to be accomplished will provide the basis for exploitation of this new route of continuous release of drugs by establishing principles for design of tubule-forming lipopeptides and other prodrugs of therapeutic value.
Recently, Mannino et al. described a lipid matrix-based approach to the oral delivery of vaccines [10-13] . Protein cochleate vaccines involve the incorporation of protein antigens into phosphatidylserine-calcium precipitates. Calcium is responsible for maintaining the cochleate structure because it interacts with negatively charged phosphatidylserines between successive bilayers. In the absence of calcium, the cochleates unroll to form liposomes. The protein cochleate approach to vaccine delivery is successful, and oral administration results in strong mucosal and systemic antibody responses, and long-term immunological memory to influenza and parainfluenza type I virus glycoproteins [12] . The structure of the cochleate protects the protein antigens from degradation in the stomach, and it is believed that the conversion to a liposomal form by the loss of calcium facilitates uptake of the antigen by the Peyer's patches of the small intestine [12] .
The lipid tubule approach to vaccine delivery is similar but has
several distinct advantages over the protein cochleate approach.
Lipid tubules do not require calcium to maintain their self-assembled
structure so they should be less sensitive to varying levels of
calcium present in the gastrointestinal tract that arise from
differing types of meals. Drug loading can potentially be much higher
in a tubule because bilayers do not have to be brought into as close
proximity in a tubule microstructure as the do in a cochleate
cylinder. Incorporation of membrane proteins and lipidated peptides
into the bilayer must disrupt the cochleate structure at least
locally. Finally, some lipidated peptides form tubules by themselves
in the absence of additional matrix lipids. If antigenic lipidated
peptides can form tubule microstructures by themselves, then antigen
loading could be much greater than the loading in a cochleate
cylinder. Gaining an understanding of
the mechanisms that contribute to tubule formation and cause
subsequent release from these microstructures will allow for the
rational and intelligent design of novel tubule delivery devices.
The following sections describe the bulk of the work performed in the last year. For context, the timeline predicted for the grant as submitted to NIH/Whitaker is included as an Appendix.
Of the two tetrapeptide Gly-Lys(e-Z)-Sar-Pro-Glu(NHC12H25)2, Gly-Lys-Sar-Pro-Glu(NHC12H25)2 and a tripeptide, (Pro)3-Glu(NHC12H25)2, study-model amphiphiles of Shimizu et. al. [5] and a new compound, Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2, the triproline head group amphiphile formed better defined helical ribbons and hollow tubules under our microstructure formation conditions (see elsewhere in this report). The tetrapeptide amphiphiles formed long cylindrical rod-like microstructures with the dimensions of 0.1 to 0.9 µm diameter x >70 µm length as observed from optical micrographs (no direct TEM or freeze fracture replica made), suggesting varying layers of bilayer rolled-up cochleate cylinders.
The triproline head group was chosen as a core peptide lipid and their variations were made on this amphiphile. First, the lipid chain lengths were increased by 2 and 4 carbons on each lipid chain to create amphiphiles with aggregation and melting properties desirable under physiological conditions. (Pro)3-Glu(NHC14H29)2 and (Pro)3-Glu(NHC16H33)2 were synthesized for the formation of microstructures and the determination of physical parameters (Tm and CAC) from MeOH and buffer at physiological pH and ionic strength. Of the three amphiphiles, (Pro)3-Glu(NHC16H33)2 gave the highest tubule yield at first trial, and thus, was chosen to label with isotopic tracer for studying distribution of the lipopeptide in mice, measurement of CAC, and of dissolution rates (see elsewhere for details). [5-3H]Pro-(Pro)2-Glu(NHC16H33)2 was prepared with the specific activity of 10 Ci/mole. The animal study, however, was not done due to the unbudgeted high cost. The CAC of the amphiphile in the formation buffer (20 mM HEPES, 120 mM NaCl, 1 mM EDTA, 0.2 % NaN3, pH 7.4) was estimated to be 5.8 ± 0.4 x 10-9 M.
The first round of peptide amphiphiles designed, containing a potential tryptic cleavage site, Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2, by making minor modifications on one of Shimizu peptide amphiphiles reported to form tubules [5], and the corresponding peptide head group alone, Ac-Lys-Ala-Sar-Pro, were evaluated as trypsin substrates. Both compounds proved to be very poor substrates, so this peptide sequence as abandonded as a substrate to drug release by proteolysis. It may prove useful as a spacer tubule forming core lipid, however.
The "second generation" peptide amphiphiles contain proven tryptic cleavage sites created by removal of the N-methylated amino acid (Sar) and lengthening of the peptide chain length to 10 amino acids. Simple modeling results from Ac-Lys-Ala-Sar-(Pro)3-Glu(NHC12H25)2 indicated a minimum of seven amino acid tether was required to prevent unfavorable steric interactions (see last year's report). Considerations of the melting temperature and CAC predictions, based on existing literature values, were also part of the design. Ac-Gly-Arg-Ala-(Gly)2-(Ala)2-(Pro)3-Glu(NHC14H29)2 was synthesized. This novel amphiphile generated microstructures of helical ribbons and hollow tubes from 30% MeOH/HEPES buffered saline, pH 7.4 (see elsewhere for details).
In preparation of the tryptic cleavage study, the peptide head group (Ac-GRAGGAAPPP, named Ac-peptide 2 from here on) without the lipid was also prepared. Both the peptide alone and the lipopeptide were subjected to tryptic hydrolysis and they resulted in complete hydrolysis in 4 hrs at 37 _C, indicating that the peptide sequence is a good substrate for trypsin. The hydrolytic reaction product Ac-Gly-Arg was also prepared independently to develop methods of detection. A number of cycles of synthesis of Ac-peptide 2 amphiphile were effected for various reasons. Different synthetic strategies were also employed. The inefficiency, inconvenience, and low yield of tritioacetylation of peptides coupled with low specific activity, existing in the literature, prompted the design of a new synthetic approach which resulted in a discovery of a new synthetic method to making high specific activity tritioacetylated peptide amphiphiles (or peptides) with increased efficiency, convenience, and higher yield.
In order to demonstrate and establish a model drug release system by enzymatic hydrolysis, tritium labeling was chosen. Since the initial hypothesis of the tryptic release of drug molecules from the tubule structure, a new direction of hypothesis has been made as the likely mode of release by tryptic hydrolysis. The major mode of tryptic release of the model drug molecule is most likely be from the dissolved monomers and not from the crystalline tubule surfaces. In other words, peptides/drugs in tubular crystalline form are hypothesized to be mostly protected from tryptic release until dissolution of the monomers. The proof of this hypothesis entails measurements of tryptic hydrolysis kinetic rates below, at, and above CAC, requiring relatively large amounts of high specific activity labeling of the compound. [3H]Ac-peptide 2-Glu(NHC14H29)2 with the specific activity of 9.3 Ci/mmole was thus prepared to measure the hydrolysis rates at dissolved monomeric lipopeptide concentrations below guessed CAC values. The protection or release of the model drug peptide from the tubular structure will also be demonstrated by these kinetic rate measurements.
The effects of sphingolipid fatty acyl chain length and unsaturation, a-hydroxylation, headgroup, and formation conditions were studied in order to understand their role in the formation of supramolecular assemblies. The results are summarized in Table I. The simplest glycosphingolipid is galactocerebroside (GalCer). GalCer can be divided into two major subfractions, that which contains a-hydroxy fatty acid (HFA-GalCer) and that which contains non-hydroxy fatty acid (NFA-GalCer).sphingosine. Removal of the sugar headgroup provides ceramide (Cer). Further removal of the fatty acyl chain to generate a 1° amine yields sphingosine.
The ability of galactocerebroside (GalCer) to form high-axial-ratio-microstructures (HARMs) has been studied by a variety of methods; evaporation of aqueous pyridine, thermal cycling in mixtures of ethylene glycol and water or phosphate buffered aqueous saline, and precipitation from dimethylformamide (DMF) by water addition. The assembly morphology of HFA-GalCer is dependent on the formation conditions. When formed by pyridine evaporation or thermal cycling, HFA-GalCer forms cochleate cylinders, whereas DMF/H2O precipitation yields hollow tubes with some helical ribbon content. On the other hand, NFA-GalCer forms very long tubules (mm) under all formation conditions. In general, DMF/H2O precipitated assemblies have smaller diameters (25 nm) as compared to other preparation methods (50-100 nm). Furthermore, the use of aqueous saline solution in place of water does not effect the tubule diameter.
Since NFA-GalCer is composed of a nonhomogeneous population of fatty acyl chains, the ability of a pure single molecular species of NFA-GalCer to form HARMs were evaluated using DMF/H2O precipitation. GalCer containing long mono-unsaturated fatty acids chains, 24:1-GalCer and 18:1-GalCer, form hollow tubules. On the other hand, long saturated fatty acid containing GalCer species, 18:0-GalCer and 16:0-GalCer, form ribbons. The physical size of pure GalCer HARMs are of a comparable to that of NFA-GalCer.
The ability of galactose containing sphingolipids to form HARMs is attributed to an inter- and intramolecular hydrogen bonding network between headgroups. In order to evaluate headgroup influence on assembly morphology, pure ceramides were investigated. As before DMF/H2O precipitation of long mono-unsaturated fatty acid containing ceramides, 24:1-Cer and 18:1-Cer, form cylindrical nanostructures whereas long saturated ceramides, 18:0-Cer and 16:0-Cer, give ribbons. Further shortening of the fatty acyl chain, 6:0-Cer, results in the formation of hollow tubes whereas 2:0-Cer forms amorphous aggregates. Furthermore, it has been shown that the amide carbonyl plays a role in assembly formation. Reduction of the 24:1-Cer amide to the 2° amine results in the formation of amorphous aggregates, whereas, psychosine, a galactocerebroside lacking the fatty acyl chain (1° amine) forms HARMs.
GalCer analogs in which the galactose headgroup was replaced with peptidyl ester linked N-acetylated amino acids were also prepared and studied (six synthetic steps from commercially available sphingosine). A twenty-four carbon fatty acyl chain with mono-unsaturation was selected as a core amphiphile due its high natural abundance in NFA-GalCer. Ceramide containing a glycine headgroup formed cylindrical HARMs when formed by DMF/H2O precipitation; however, proline containing ceramide (Pro-Cer) would not form assemblies using any of the previously mentioned conditions. This unexpected result, in light of the success of the glutamine dialkyl amides with proline headgroups, led to the study of mixed lipid systems.
Various intimate mixtures of Pro-Cer with either NFA-GalCer or 24:1-GalCer in DMF were prepared and then the HARMs generated by water addition. The morphology of the mixed lipid assemblies are similar to those of the pure matrix amphiphile (hollow tubes). Increasing the proline component above 25% results in the formation of vesicles in addition to tubules. Nuclear magnetic resonance analysis of the isolated tubules (25 mole% NAcPro-24:1-Cer) reveal that the precipitated HARMs retain the original solution composition. Furthermore, tubule morphology remains unchanged when suspended in a pure aqueous environment. Differential scanning calorimetry studies indicate that the lipids are ideally mixed.
Due to the inherent instability and synthetic limitations imposed by a peptidyl ester linkage, 24:1-Cer's 1° alcohol was converted to the corresponding amine (seven synthetic steps from sphingosine). This will allow for a greater diversity of amino acid headgroups used in HARM studies. More importantly, the amine provides a handle by which further chemical modifications (i.e. bioconjugation to drugs, peptides, and fluorescent probes) can occur in aqueous media and after the HARM has been formed.
Gastrointestinal administration of peptide drugs is a worthy objective but difficult to achieve in practice. When administered orally, peptides are usually degraded by the enzymatic environments of the gastrointestinal tract and especially by the acidic environment of the stomach. In addition, water soluble peptide drugs are poorly absorbed. It is generally agreed that less than 1% of an orally administered does of peptide drug finds its way into the systemic circulation [14, 15]. The self-assembly of hydrophobically-modified peptide drugs into lipid tubule microstructures offers several potential advantages to oral drug delivery in that the tight crystalline packing of the tubule offers stability and protection.
Bile salt surfactants, along with other components of biliary secretion including phospholipids, cholesterol, triglycerides, pigments, and ions, all interact with surface active drugs. They play an important role in fat emulsification and absorption within the small intestine. For liposomal drug delivery, it is well known that the interaction between bile salt surfactants and phospholipid bilayers affects the physical state and integrity of liposomes [16, 17]. Certain solutes directly affect the phase transitions of phospholipids by altering the rigidity of the bilayer membrane, and in extreme cases liposomes may be destroyed and converted to mixed micelle systems by the presence of surface-active agents [18].
Except at high dilution, detergent solutions contain micelles, which are thermodynamically stable small aggregates roughly 4 nm in diameter. Micelles differ from lamellar bilayer aggregates not only in size but also in structure. Micelles have a water soluble, hydrophilic surface and a hydrophobic core. When lamellar bilayer vesicles are exposed to a detergent, an equilibrium state is approached quickly as detergents partition into the bilayer [19]. Because of membrane packing considerations, the incorporation of detergent molecules into a bilayer destabilizes the bilayer structure [20]. Transformation of all the lamellar vesicles into mixed micelles occurs when the effective ratio of detergent to lipid exceeds a critical value [21].
The tight crystalline packing of a lipid tubule provides protection for covalently-bound peptide drugs while in the acid environment of the stomach, but the presence of bile in the small intestine and its ability to solubilize bilayer membranes provide a mechanism to release drugs or antigens from a tubule delivery device. An understanding of the mechanisms and kinetics for the solubilization of tubule microstructures by detergents and bile acids is a critical first-step toward the design of gastrointestinal-based delivery systems. The design of an oral delivery vehicle involves some important considerations. First, the labile peptide drug and its delivery system both must be stabile within the harsh acidic environment of the stomach. Second, the delivery system must be designed to degrade fully while the device is in the small intestine because the transit time of the delivery vehicle within the GI-tract is small and finite. The experiments described within this section explore the stability of Pro3-Glu-(NC16)2 tubules in acidic environments (akin to the low pH of the stomach) and investigate the solubilization kinetics of DC8,9PC tubules by the synthetic non-ionic detergent octylglucoside.
Solubilization by Surfactants
Studies on the interactions of surfactants with lipid vesicles have led to the use of synthetic lipid vesicles as models for biological membranes, and many of the kinetic studies on membrane solubilization by detergents focus on the extraction of membrane proteins and the preparation of unilamellar vesicles [18]. Within the drug delivery community, however, solubilization by surfactants is an important consideration for gastrointestinal drug delivery. The interactions of bile salts and other biological surfactants have been investigated to enhance the solubility of poorly soluble particulate drugs [22, 23]. Bile salts and other digestive components, such as fatty acids, monoglycerides, and phospholipids, that are dissolved in biliary secretions have been examined in detail because of their effects on solubilization, dissolution, and absorption within the small intestine [24]. For most bile salts, the concentration within the lumen of the intestine is about 10-fold higher than the critical micelle concentration [25].
The effect of the detergent-to-phospholipid ratio on the solubilization of large, unilamellar phosphatidylcholine vesicles by a variety of detergents has been well-described by a three stage model, where the amount of detergent considered is that in excess of the CMC [26, 27]. Solubilization of bilayer vesicles generally occurs above the CMC of the detergent; for a lipid to leave the bilayer surface in a mixed micelle, the micelle must be thermodynamically stable. At low detergent to phospholipid ratios (i.e. < 1.5), the detergent distributes between the bilayer and the aqueous solution with a partition coefficient of about 60. The lamellar bilayer rearranges structurally to accommodate the presence of the detergent but does not solubilize. When the detergent to lipid ratio exceeds a critical value (i.e. 1.5 - 3 depending on the system), the detergent-saturated bilayers convert into mixed micelles. At high detergent to lipid ratios (> 3), lamellar bilayer structures are completely solubilized. As the ratio increases, micelles become smaller in size and contain less phospholipid.
Partitioning of the detergent into the bilayer and exchange of phospholipids between mixed micelles are fast processes. It has been proposed that the lamellar-to-micelle transition is the rate limiting step in the solubilization process [19]. The solubilization of egg phosphatidylcholine by the bile salt sodium deoxycholate occurs by a first-order mechanism, which implies that micellization is stochastically-driven process that depends on the amount of exposed bilayer surface [28, 29].
Preparation of Pro3Glu(NC16)2 Tubules and Protection against Acid Hydrolysis
Tubules composed of Pro3Glu(NC16)2 lipids were prepared by precipitation upon dilution of concentrated MeOH solutions of the compounds with water. Samples of either 0.1 mg of Pro3Glu(NC16)2 dissolved in 20 µl of MeOH were added to 200 µl of water while vortexing. Sealed samples were heated to 65 °C, incubated for 15 min., then cooled to room temperature at 0.2 °C/min.
For studies of Pro3Glu(NC16)2 stability to acid hydrolysis, a 0.5 ml aliquot of an aqueous tubule suspension (pH 7.0) was placed into a micro-dialysis tube, which was created from a modified 1.5 ml Eppendorf tube with a small hole drilled into its cap. After addition of the suspension, the open end of an Eppendorf tube was covered with a small piece of dialysis tubing, and the cap was closed. The Eppendorf tube was placed into an acidic dialysis exchange solution (pH 3.0). After 16 hrs of solvent exchange by dialysis, the sample was removed from the acid solution. Prior to any microscopic examination, the pH of the suspension was measured, and a fraction of the suspension was spotted on a TLC plate.
Preparation of DC8,9PC Tubules and DPPC Vesicles
Both DC8,9PC and octylglucoside have spectral properties that make them convenient to use for monitoring tubule solubilization. DC8,9PC was obtained from Avanti Polar Lipids (Birmingham, AL) as a dry powder. Thin layer chromatography (TLC) using chloroform:methanol:water (65:35:5) showed no hydrolysis product spots, and the lipid was used without subsequent purification. Suspensions of DC8,9PC tubules were prepared by precipitation of the solvated lipid from ethanol with drop-wise addition of water (Georger, et al., 1987). Deionized water was added to a 5 mM solution of the lipid in ethanol until the volume fraction of water in ethanol reached 70%; precipitation occurred, and the solution became turbid. The microstructures were allowed to anneal in the 70% ethanol solution for one week at room temperature. Ethanol was removed by a series of centrifugation and resuspension steps in distilled water. For the final exchange, the pellet of tubules was resuspended in isotonic digestion buffer (10.0 mM Tris-HCl, 10 mM CaCl2, 150 mM NaCl, and 0.01 weight% sodium azide, pH 8.5).
DPPC was obtained from Avanti Polar Lipids (Birmingham, AL) dissolved in chloroform. TLC showed no hydrolysis product spots, and this lipid was used without purification. DPPC was dried from chloroform to create a 5 mM stock solution upon resuspension with the isotonic digestion buffer. Lipids were suspended in deionized, distilled water, and the DPPC vesicle suspension remained milky and turbid after sonication, which suggests that it contained a mixture of vesicle microstructures.
DC8,9PC Solubilization Kinetics by Octylglucoside
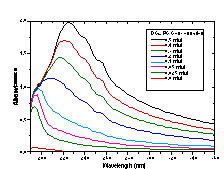
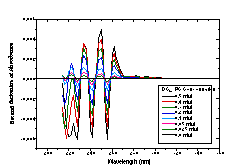
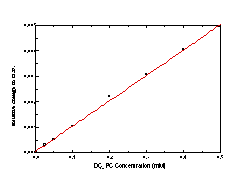
The time course of solubilization of DC8,9PC tubule microstructures by octylglucoside (OG) was monitored by measuring the intensity of the distinctive DC8,9PC absorption bands (from 200 to 260 nm) in the micellar detergent phase. Unfortunately, these suspensions are turbid in the UV, and their spectra have severe scattering (Figure 4A). To circumvent this scattering problem, a calibration curve for DC8,9PC in the presence of OG was constructed not from the raw absorption spectra but from the second derivative of the spectra (Figure 4B). To construct the calibration curve, known amounts of DC8,9PC tubules were added to aqueous OG solutions to produce final 50 mM OG concentrations. The detergent/phospholipid suspensions were heated to 65 °C to disperse the lipid into detergent micelles. The clarified solutions of mixed micelles were cooled to room temperature before being placed in quartz cuvettes to collect spectra using a Hewlett-Packard 8452A diode array UV-Vis spectrometer. Figure 4C shows the calibration curve created by plotting the value of the second derivative at 250 nm as a function of DC8,9PC concentration.
To measure the solubilization kinetics, at time 0 an aliquot of the 5 mM stock DC8,9PC tubule suspension was added to a stirred solution of OG (Sigma Chemical, St. Louis, MO) in distilled, deionized water. The final aqueous suspension had a 0.5 mM DC8,9PC concentration and a 50 mM OG concentration. Throughout the course of solubilization, the suspension was held at room temperature (~ 21.5 °C). Every 10 minutes, a 1 ml aliquot was removed from the suspension by a syringe. The aliquot was passed through a 0.2 mm Whatman Anotop 10 sterile syringe-type filter to separate micelles (~ 3-4 nm diameter) from tubules ( >500 nm diameter). The filtrate was collected in a small volume, 1 cm path length quartz cuvette, and the absorption spectrum of the filtrate was taken using a Hewlett-Packard 8452A diode array UV-Vis spectrometer. The second derivative of the spectrum was calculated, and the concentration of DC8,9PC within the micellar phase was determined from the calibration curve in Figure 4C.
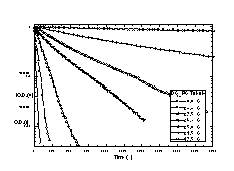
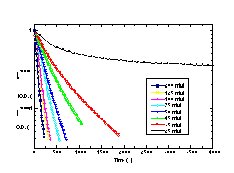
During this solubilization experiment, it was observed that the optical density at 400 nm correlated well with the amount of DC8,9PC remaining within the tubule bilayer phase. To test this correlation, a second experiment was performed in which 2.5 ml of OG in distilled, deionized water was added to a standard, 1 cm path length quartz cuvette. The cuvette was stirred and held at 21.5 °C within the UV-Vis spectrometer. Like before, an aliquot of the stock DC8,9PC suspension was added to the aqueous OG solution at time 0. The optical density at 400 nm was measured at 60 s intervals using the computer software analysis package that controlled the UV-Vis spectrometer. Figure 5 shows that rates of solubilization for long periods of time after addition of tubules to detergent were similar if determined by using optical density measurement or by the filtration technique. Measurement of solubilization using optical density was easier to implement and allowed for the measurement of faster kinetic processes.
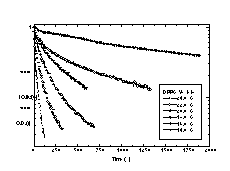
Solubilization rates were determined for DC8,9PC tubules and DPPC vesicles as a function of temperature using optical density. Each stirred cuvette of OG solution was allowed to reach thermal equilibrium prior to the addition of DC8,9PC or DPPC. Solubilization rates were measured for DC8,9PC at seven temperatures between 17.5 and 30.0 °C, and solubilization rates were measured for DPPC at six temperatures between 14.0 and 24.0 °C. Rates and temperatures were used to calculate activation energies for solubilization.
Similarly, turbidity was used to determine solubilization rates OG detergent concentration at a fixed DC8,9PC concentration (0.5 mM) and temperature (26.3 °C). Solubilization rates were measured at eight OG concentrations that ranged between 25 mM and 200 mM. The effect of OG concentration on solubilization rate was used to determined a second order rate constant for solubilization.
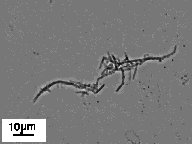
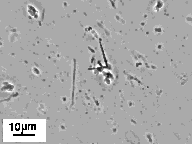
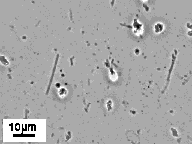
The micrographs in Figure 6 show little change in the tubule morphology upon a 12 hr exposure to an acidic environment. Although the tubule microstructure is stable at pH 3.4, some hydrolysis of the peptide did occur. The appearance of two new spots on TLC (e.g. in addition to the pure P3E-(NC16)2 spot) confirms modest degradation of the lipid upon exposure to an acidic environment.
|
|
|
Figure 6. Phase-contrast optical micrographs showing tubule and helical ribbon microstructures formed by Pro3Glu(NC16)2 in (A) physiologic (i.e. pH 7.4) and (B) acidic (i.e. pH 3.4) environments. Hydrolysis of the lipid occurs upon exposure to an acidic environment, but the morphology remains unchanged.
From a more general approach, the solubilization of phospholipid microstructures by detergents can be described by the following second-order reaction:
where [ L ] is the concentration of lipid, [ D ] is the concentration of detergent in the form of micelles (e.g. above the CMC), and [ M ] is the concentration of mixed micelles. It has been proposed that transfer of phospholipid among micelles is very rapid so that all micelles can be considered as being identical [19]. If this assumption is correct, then the reaction can be re-written as a pseudo-first order rate equation:
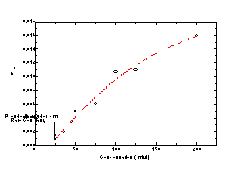
where keffective is the effective rate constant for the pseudo-first order reaction. A 100:1 molar ratio of detergent to phospholipid is well in excess of that required to cause solubilization of phospholipid vesicle membranes [21], and a tubule microstructure is no different in this regard. The data plotted in Figure 2 and show that the process of tubule solubilization proceeds by a first order mechanism. Interestingly, this is consistent with what has been reported in the literature for phospholipid vesicle microstructures [19].
The nature of the molecular and supramolecular mechanisms behind membrane solubilization are still poorly understood, but the following pictorial representation (Figure 7) encompasses all that is currently known about the process. Experiments from turbidity, NMR, and fluorescence anisotropy studies of phospholipid vesicle solubilization show that at low detergent concentrations an equilibrium established quickly between detergent in the aqueous phase and the bilayer phase [21, 30]. The vesicle swells, and the membrane becomes more fluid to accommodate the insertion of detergent, but the lamellar-to-micellar transition does not occur until a critical concentration of detergent has been exceeded. In the presence of excess detergent, the homogeneously saturated membrane begins to form micelles. Restructuring of the bilayer to form a micelle is the rate-limiting step. As long as the detergent concentration is large enough the support the formation of mixed micelles, solubilization continues. Interestingly, the solubilization of vesicle and tubule membranes would be indistinguishable because the rate depends upon the amount of bilayer surface and the stochastically-driven micellization step.
Figure 4. A model for the solubilization of lamellar bilayers by detergent micelles. The partitioning of detergent into the bilayer and the rearrangement of phospholipids between micelles are fast processes. The rate determining step is the lamellar-to-micelle transition that occurs prior to the removal of phospholipid by a mixed micelle
To confirm that multilamellar tubule and multilamellar vesicle microstructures solubilize with the same order kinetics, the temperature dependence of DC8,9PC tubule solubilization was compared to the temperature dependence of DPPC vesicle solubilization. Both phospholipids have similar Tms, are crystalline at the temperatures studied, and should have similar degrees of membrane fluidity. Figure 8 shows the change in the optical density of the detergent/phospholipid suspensions as a function of time. The shapes of the solubilization profiles reflect that DC8,9PC tubule solubilization is a single exponential, first-order process at all temperatures. However, careful inspection shows that DPPC vesicle solubilization proceeds by a multi-exponential, first-order process. Many phenomena affect turbidity, including changes in concentration, size, and shape of scattering objects. Based on a large body of fluorescence anisotropy measurements, it has been proposed that large vesicles first go to an intermediate small vesicle structure of a critical size prior to the lamellar-to-micelle transition [30]. The initial, fast loss in turbidity may reflect a change in vesicle size while the slow loss may reflect the lamellar-to-micelle transition.
|
|
|
The Arrhenius plot in Figure 9 shows the effect of temperature on
the solubilization rate for tubules and vesicles from which
activation energies of solubilization were calculated. In the case of
DPPC solubilization, the rate constant was determined from the
optical density data after the initial fast rate. The energy barrier
for solubilization of a DC8,9PC tubule
microstructure is modestly higher at 369 kJ mol-1 than for a DPPC
vesicle at 270 kJ mol-1. This discrepancy can be explained in part by
the higher melting temperature for DC8,9PC
(43.8 °C) compared to DPPC (41.3 °C). Interestingly, the
intrinsic solubilization rate of DPPC is about 25 times faster than
that of DC8,9PC at room temperature (21.5
°C). The molecular rearrangements required for the formation of
a mixed micelle from a DPPC vesicle must be less stringent than those
for a DC8,9PC tubule.
|
|
Figure 9. Arrhenius plots of solubilization rates for DC8,9PC tubule and DPPC vesicle suspensions. Rate constants were determined for the solubilization of suspensions that had 0.4 mM phospholipid and 40 mM OG. |
|
A |
B |
The first-order solubilization profile of self-assembled tubule microstructures suggests that this model delivery system may function as a controlled drug or vaccine delivery device when administered perorally. These results demonstrate that even with a tightly packed crystalline microstructure, lipid tubules can be solubilized by detergents. This form of chemical delivery has great potential and may intrinsically allow for the protection of labile peptide drugs from premature hydrolysis in the gut due to the tight molecular packing of the crystalline bilayer. As was demonstrated in the interfacial enzymatic hydrolysis studies, only the lipids present at phase boundaries and crystal edges, like those at tubule ends, would be accessible to enzymatic attack. Peptide drugs may then survive the acidic and enzymatic environments of the stomach on their way to the small intestine.
Mechanistically, solubilization is a completely different release process than interfacial enzymatic hydrolysis even though both mechanisms require the presence of an additional facilitator (e.g. either detergent micelle or interfacial enzyme). However, they appear to differ in how the facilitator interacts with the bilayer. Detergents disrupt bilayer packing and solubilize lipid from surfaces because they can intercalate into the membrane from anywhere on the bilayer surface. Interfacial enzymes, like PLA2, predominantly act at defects and phase boundaries in crystalline bilayers. In effect, these kinetic studies support the theory that enzymatic hydrolysis works at edges and micellization acts at surfaces. Dissolution, which is the third mechanism of release to be studied in this proposal, should be zero-order. Unlike enzymatic release, dissolution does not require any facilitators. Drugs are released by the loss of material from an eroding bilayer edge. The experiments described within the following sections outline how this important release mechanism will be explored.
Pure compounds
The values of phase transition temperatures (Tm) must be known to develop protocols for forming, storing, and observing HARMs. To determine these values, differential scanning calorimetry (DSC) of the compounds of interest was performed. To prepare samples for DSC, 70 µl of an appropriate aqueous medium was added to 1.0-2.0 mg of lipid powder. The samples were sonicated for 5-30 min. at room temperature using a water bath sonicator (Laboratory Supplies Co., Hicksville, NY). The resulting suspensions were sealed in 70 µl silver calorimetry pans in aliquots containing 50 mg of aqueous medium. A pan of the same type with 50 mg of pure aqueous medium was used as a reference. DSC was performed using a Seiko DSC-100 high sensitivity calorimeter. Samples were heated from 3 °C to 95 °C at 1 °C/min. In order to insure proper hydration of the peptide lipids, each sample was pre-heated under the same conditions before the final calorimetric scan. To control integrity of the compounds, after DSC the pans were opened and their contents were extracted with appropriate organic solvents. The resulting extracts were analyzed by TLC. TLC revealed no decomposition of the studied compounds after DSC.
The phase transitions obtained are summarized in Table II, which shows that all compounds tested except (Pro)3-Glu(NHC12H25)-NHC12H25 and 24:1-amine have Tm values higher than 37°C. Since all HARMs observed to date are stable below their lipid chain melting temperature, particles formed of the compounds studied here should be stable at physiological temperature.
|
|
|
|
|
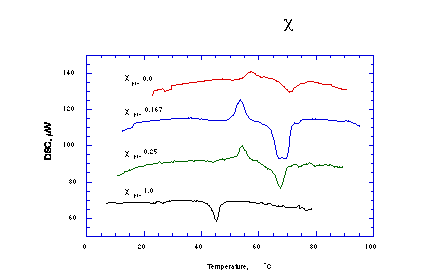
DSC was also employed to determine if Pro-24:1-Cer and NFA-GalCer are uniformly mixed in the HARMs formed from these two amphiphiles using DMF/H2O precipitation method. In this case, suspensions of pre-formed HARMs containing 0.5-1 mg of total lipids in 50 µl of H2O were thermally scanned from 5 to 95°C at 1°C/min. The traces obtained were normalized to 0.2 mg of Pro-24:1-Cer (Figure 11). The fact that both Pro-24:1-Cer/NFA-GalCer 1/5 M/M (Figure 11, c=0.167) and Pro-24:1-Cer/NFA-GalCer 1/3 M/M (Figure 11, c=0.25) HARMs lack phase transition characteristic for pure Pro-24:1-Cer (Figure 11, c=1) clearly indicates that the components were uniformly mixed. The presence of exotherms before the main NFA-GalCer transition may be due to previously reported metastable states [32, 33]. In pure NFA-GalCer, these metastable states are related to the hydration state of the sphingolipid.
|
|
Figure 11. Differential scanning calorimetry heating curves of Pro-24:1-Cer : NFA-GalCer mixed lipid systems. |
The stability of lipopeptide HARMs in any solution is controlled by the critical aggregation concentration (CAC--that concentration at which the first microstructures form from a solution of monomers), since at equilibrium there will always be monomers at the CAC present in solution. A high value of a lipopeptide CAC could lead rates of microstructure dissolution too high for practical use in drug delivery systems. Also, subsequent studies of enzymatic degradation of HARMs require must determine whether enzymes act on the soluble monomers or on lipopeptides within the microstructures themselves.
CAC of a representative -C12H25 lipopeptide, Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2, was measured as concentration of its saturated solution. An 0.5 ml aliquot of 1 mg/ml Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2 solution in MeOH was dried by N2 flow at room temperature and for 2 hours under oil-pump vacuum at room temperature consequently. The dried lipidated peptide was incubated in 10 ml of HBS overnight at room temperature with shaking. Dissolved fraction of the lipidated peptide was isolated by centrifuge driven filtration. To do this, 1.5 ml aliquots (in triplicate) of the resulting dispersion were centrifuged on Ultrafree®-CL Centrifugal Filters with 30,000 Da nominal molecular weight limit (Millipore Co., Bedford, MA) for 15 min. at 3000 x g at room temperature. Aliquots of 1 ml of the resulting filtrates were withdrawn. After discarding of the excess of the filtrates, each filtrate cup was washed with 0.5 ml of 10 % Triton X-100 in 100 mM Na+-borate buffer pH 9.0. Triton washes were pooled with appropriate Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2 filtrates. Concentrations of Ac-Lys-Ala-Sar-Pro- Glu(NHC12H25)2 in the resulting samples were determined by fluorescamine methods adopted from the technique suggested by Bohlen et al. [34]. Briefly. Aliquots of 0.5 ml of freshly dissolved 0.3 mg/ml fluorescamine in dioxane were added to the samples while vortexing. The resulting mixtures were incubated for 30 min. Fluorescence of the samples were measured at 390 nm excitation, 475 nm emission wavelengths using Perkin-Elmer LS-5b Luminescence Spectrometer. Concentrations of Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2 were determined using calibration curve. The calibration curve was constructed using 0-2.6x10-9 nmole samples of Ac-Lys-Ala-Sar-Pro- Glu(NHC12H25)2 solubilized in 1.5 ml of 6.7% Triton X-100 in 100 mM Na+-borate buffer pH 9.0 and mixed with 0.3 mg/ml fluorescamine in dioxane at the same time as analyzed filtrates. CAC of Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2 was found to be 2.2 x 10 + 0.3 x 10-6M (n=3).
Unfortunately not all synthesized lipopeptides has the free NH2-group in the composition. Moreover, analysis of literature on CAC of phosphatidylcholines with various hydrocarbon chain length [35] shows that sensitivity neither of the fluorescamine method nor other physico-chemical approaches, such as incorporation of lipophilic fluorescent probes into micelles or surface tension, most probably, would be sufficient for determination of CAC of most promising -C14 and -C16 lipopeptides. Because of this, we decided to synthesize radiolabeled analogous of lipopeptides. This is most sensitive, but very expensive and time consuming method for concentration measurement. So, only synthesis of [5-3H](Pro)3-Glu(NHC16H33)2 and consequent CAC measurement has been accomplished to date.
Preparation of a saturated solution and separation procedure applied in the case of [5-3H](Pro)3-Glu(NHC16H33)2 were close to that of Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2. An 0.5 ml aliquot of 1 mg/ml [5-3H](Pro)3-Glu(NHC16H33)2 solution was dried by N2 flow and under oil-pump vacuum consequently. The dried lipidated peptide was incubated in 50 ml of HBS overnight at room temperature with shaking. The resulting dispersion was centrifuged for 30 min. at 10000 rpm 25°C. Forty eight ml of the supernatant was discarded. Fresh HBS was added to the pellet in residual 2 ml of the supernatant to the final volume of 40 ml. The resulting suspension was incubated at room temperature with shaking. Aliquots of 10 ml were withdrawn on the 1st, 2nd, and 3d day of incubation. Dissolved fraction of the lipidated peptide was isolated by centrifuge driven filtration. The suspensions from the sample reservoir and the rest of the filtrates were removed, and 2 other portions of the fresh suspension were filtered using the same filtration units consequently. For each time point, the filtrations were done in duplicates. Radioactivities of 1 ml of the resulting filtrates were determined after mixing with 15 ml of liquid scintillator using ß-counter. Concentrations of the (Pro)3-Glu(NHC16H33)2 in the filtrates were calculated using specific radioactivity value.
This method for CAC measurement appears successful, since CAC values obtained on all three days are constant, and since there was no difference between sequential filtrations through the same filter, adsorption on the filter must have been negligible. The CAC value of (Pro)3-Glu(NHC16H33)2 was found to be 5.8 ± 0.4 x 10-9 M (n=2); this value is not inconsistent with CAC values for similar compounds with shorter hydrocarbon chains reported by Shimizu et al. [5]. This value of CAC is only slightly greater than that of DPPC (4.6 x 10-10 M), and not so high that one would expect rapid dissolution of HARMs of this or similar compounds even after considerable dilution of the particles.
An ability of the compounds to form high ratio particles (HARMs) is a key property we are looking for in the current project. It was shown by Shimizu et al. that sonicated dispersions of some peptides lipidated with glutamic acid dialkyl amides (lipopeptides) are capable of forming HARMs spontaneously in pure water through "...gradual change from a very small particle to another specific morphology over a period of hours or days..." [5]. One of our primary goals was to obtain similar particles in a much shorter time. To this end, we applied methods that form HARMs from one of the most studied tubule forming compound, the phospholipid DC8,9PC to some reported by Shimizu et al. and newly synthesized lipopeptides. The methods used included thermal cycling of aqueous dispersions [8, 36-39], and of aqueous dispersions in the presence of MeOH or EtOH [40].
When MeOH or EtOH were used in HARM preparation, the particles were subsequently transferred to pure HBS using centrifugal-driven filtration. To do this, 200 µl aliquots of particle suspensions were centrifuged in Ultrafree®-CL Centrifugal Filters with a 30,000 Da nominal molecular weight limit for 15 min at 3000 x g at room temperature, washed with 1 ml of fresh HBS under the same conditions, and reconstituted in the original volume of HBS.
The morphology of the particles was studied using a Zeiss ICM-405 inverted phase contrast microscope (Carl Zeiss, Thornwood, NY) equipped with a Dage 66 SIT video camera (Dage-MTI Inc., Michigan City, IN). Images obtained were digitized using a Data Translation QuickCapture frame grabber board in a Macintosh II. In addition, the particles were studied by transmission electron microscopy (TEM). Samples for TEM were prepared by applying 20 µl aliquots of particulate suspensions to Formvar-coated 150 mesh copper TEM sample grids. The samples were dried in air for at least 24 hr before observation with a Philips EM 410 transition electron microscope.
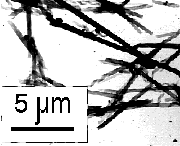
The first approach tested for quick and simple lipopeptide HARM formation was heating and cooling of aqueous dispersions of the surfactant through Tm. Dispersions of (Pro)3-containing amphiphiles with different hydrocarbon chain lengths were heated to 65 °C in HBS and cooled to room temperature at 0.2 °C/min. Samples of 0.5 mg of either (Pro)3-Glu(NHC12H25)2 or (Pro)3-Glu(NHC14H29)2 or (Pro)3-Glu(NHC16H33)2 in 1 ml of HBS each sonicated for 15 sec at room temperature using a water bath sonicator were used in these experiments. Heating and slow cooling through Tm transforms (Pro)3-Glu(NHC12H25)2 into helical ribbons with nearly 100% efficiency. The width of these ribbons is similar to the helical pitch, but fusion of adjacent ribbon edges is observed in rare cases (Fig. 12, Table III).
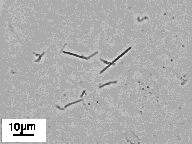
Slow cooling in HBS resulted in HARM formation by (Pro)3-Glu(NHC14H29)2 and (Pro)3-Glu(NHC16H33)2 as well. However, the conversion efficiencies in these cases were much lower; significant fractions of the material formed amorphous aggregates. In attempts to increase the conversion efficiencies through solubilization of the lipid at elevated temperatures, we supplemented the (Pro)3-Glu(NHC14H29)2 and (Pro)3-Glu(NHC16H33)2 dispersions with MeOH in various concentrations. The influence of MeOH on HARM formation has been studied previously with DC8,9PC. It has been shown that cooling of DC8,9PC dispersions in the presence of alcohols can result in more efficient tubule formation than that found with pure aqueous dispersions [40], and data obtained on (Pro)3-Glu(NHC14H29)2 and (Pro)3-Glu(NHC16H33)2 are consistent with this finding. Slow cooling of lipopeptides in the presence of 30% or 50% MeOH increases the efficiency of HARM formation to almost 100%. It should be noted that only the particles formed in 30% MeOH are relatively straight and uniform in size (Figure 12, Table III). Slow cooling in 50% MeOH induced formation of ribbons with broad width variation; some ribbons were tapered, and others were irregularly wound into imperfect helices (data not shown). Thermal cycling in MeOH in concentrations of less than 30% resulted in only partial conversion to HARMs (data not shown).
Heating and cooling of DC8,9PC aqueous dispersions in the presence of EtOH induces tubule formation as well [40]. This approach for formation of HARMs was applied to some of the lipopeptides. A single concentration of EtOH was used for each compound--that concentration at which the lipopeptide precipitates from solution in absolute EtOH upon gradual addition of HBS at room temperature. Heating to 50 °C and fast subsequent cooling (50 °C to 24 °C within about 4 min) of the resulting suspensions produced HARMs from both Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2 and Gly-Lys-Sar-Pro-Glu(NHC12H25)2. Both compounds formed mixture of very long (> 70 µm) and sometimes bent rod-like aggregates with rather broad variations in diameter (Figure 12, Table III). Cooling at a slower rate, in this case, did not significantly alter the morphology of the particles (data not shown). The rest of the compounds processed this way, Pro-Glu(NHC12H25)2 and (Pro)3-Glu(NHC12H25)2, did not form HARMs (data not shown).
Finally, we found well defined HARMs formed from Gly-Lys-Sar-Pro-Glu(NHC12H25)2, (Pro)3-Glu(NHC12H25)2 and Ac-peptide2-Glu(NHC14H29)2 just after addition of concentrated solutions of these lipopeptides in MeOH to HBS - the initial step in preparation of the lipopeptide suspensions in HBS/MeOH mixtures. Gly-Lys-Sar-Pro-Glu(NHC12H25)2 forms helical ribbons (Figure 12, Table III), (Pro)3-Glu(NHC12H25)2 forms HARMs with morphology and dimensions very close to those of obtained by slow cooling (Table III), and Ac-peptide2-Glu(NHC14H29)2 forms the tubules (Fig. 12, Table III).
|
|
|
|
|
|
|
(Pro)3-Glu(NHC12H25)2 |
Slow cooling of HBS dispersion Dilution of MeOH solution |
Helical ribbons/tubules Helical ribbons/tubules |
7.0 ± 2.5 (n=10) 6.6 ± 2.3 (n=7) |
0.14 ± 0.05 (n=7) 0.15 ± 0.04 (n=9) |
|
(Pro)3-Glu(NHC14H29)2 |
Slow cooling in 30% MeOH in HBS |
Helical ribbons/tubules |
7.8 ± 2.9 (n=7) |
0.22 ± 0.02 (n=5) |
|
(Pro)3-Glu(NHC16H33)2 |
Slow cooling in 30% MeOH in HBS |
Helical ribbons/tubules |
7.9 ± 3.2 (n=7) |
0.28 ± 0.03 (n=5) |
|
Ac-peptide2-Glu(NHC14H29)2 |
Dilution of MeOH solution |
Tubules |
7.7 ± 3.2 (n=6) |
0.40 ± 0.02 (n=5) |
|
Gly-Lys-Sar-Pro- |
Dilution of MeOH solution Cooling in 42% EtOH in HBS |
Helical ribbons
|
3.1 ± 0.3 (n=7) >70 |
0.12 ± 0.02 (n=5) 0.65 ± 0.18 (n=9) |
|
Ac-Lys-Ala-Sar-Pro- |
Cooling in 46% EtOH in HBS |
Rod-like aggregates |
>70 |
0.91 ± 0.39 (n=8) |
|
|
|
|
|
|
|
|
|
 VII VII |
Figure 12: HARMs formed from peptides lipidated with glutamic acid dialkyl amides: (Pro)3-Glu(NHC12H25)2 slow cooled in HBS (I); (Pro)3-Glu(NHC14H29)2 (II), and (Pro)3-Glu(NHC16H33)2 (III) slow cooled in 30% MeOH in HBS; Ac-Lys-Ala-Sar-Pro-Glu(NHC12H25)2 (IV), and Gly-Lys-Sar-Pro-Glu(NHC12H25)2 (V) cooled in 46% and 42% EtOH in HBS; Gly-Lys-Sar-Pro-Glu(NHC12H25)2 (VI), and Ac-peptide 2-Glu(NHC14H29)2 (VII) obtained by dilution of concentrated MeOH solutions with HBS. |
It is important for potential pharmaceutical applications that lipopeptide HARMs not change morphology due to interaction with biological fluids or cell membranes, whether through loss of monomeric surfactants or other mechanisms. (Pro)3-Glu(NHC16H33)2 was chosen to test the survival of lipopeptide HARMs in simulated biological fluids. A suspension of (Pro)3-Glu(NHC16H33)2 HARMs was formed by heating and cooling of the lipopeptide in 30% MeOH in HBS. The resulting particles were transferred to pure HBS using centrifuge driven filtration (see above). The suspension obtained was added to either fetal calf serum or sonicated DOPC liposomes in HBS at pH 7.4 at a DOPC concentration of 5 mg/ml in a ratio of 1/10 (v/v). The resulting suspensions were incubated for 45 hr at 40 °C.
|
|
| |
|
|
| |
|
|
| |
|
|
| |
(Pro)3-Glu(NHC16H33)2 was chosen for toxicity study as well. Again, particles were formed by slow cooling of the lipopeptide dispersion in 30% MeOH in HBS. In this case, the procedure was scaled up and adopted to sterile conditions. A sample of 2.8 mg of (Pro)3-E(C16H32)2 was dissolved in 1.68 ml of MeOH. Two 0.84 ml portions of the resulting solution were added slowly to two separate 1.96 ml portions of filter sterilized HBS (without EDTA and NaN3) pH 7.4 while vortexing. The resulting suspensions was heated to 65°C, and cooled down slowly (65 to 22°C in about 4 hours) in the closed test tubes. Upon cooling, the obtained particles were spun down for 20 min. at 2000xg at room temperature. Supernatants were withdrawn (2.1 ml from each sample) and substituted with 5.5 ml of fresh HBS. The centrifugation was repeated, and 5.5 ml supernatants were withdrawn again. Resulting suspensions of washed particles were pooled, and the total volume was adjusted to 3.5 ml. The obtained tubule suspension were distributed to 1 ml glass vials (Sun International) with scepter screw caps in 250 µl aliquots. Before filling of each vial the suspension was stirred by pipetting for 5 times. The control samples were prepared in exactly the same way as the tubule suspension, but instead of (Pro)3-E(C16H32)2 solution, pure MeOH was used. In order to obtain sterile samples, all manipulations described above that involved addition or withdrawal of solutions were performed in the laminar flow hood using steam sterilized glassware and sterile disposable plastic pipettes. The morphology of particles obtained was studied using optical microscopy. It was shown that changes in technique due to adaptation to sterile conditions did not affect morphology of the resulting (Pro)3-E(C16H32)2 particles (data not shown).
In order to measure the final concentration of (Pro)3-E(C16H32)2 in the resulting suspension, the content of one of the vials (250µl) was freeze-dried, and (Pro)3-E(C16H32)2 was extracted into 250 µl of MeOH. MeOH extracts of freeze-dried HBS samples containing known amounts of (Pro)3-E(C16H32)2 were used as standards. Eight µl of the extracts were analyzed by TLC in But-OH/Ac-OH/H2O, 4/1/1 v/v/v. Spots were stained with 1% ninhydrin solution in 100/3 v/v mixture of But-OH/Ac-OH. The stained TLC plate was digitized, and areas, and average intensities of the spots were measured using image-processing program NIH Image. The concentration of (Pro)3-E(C16H32)2 in the tubular suspension was calculated using a calibration curve constructed using average intensities and areas of the spots with known concentrations. Obtained value of 0.6 mg/ml of (Pro)3-Glu(NHC16H33)2 corresponds to 75% overall yield of the tubule preparation procedure.
Biosupport, Inc. conducted a dosage-range evaluation of the (Pro)3-Glu(NHC16H33)2 HARMs Male Balb C mice, 18-22 g, 3-12 months old were injected with either 250 µl (150 µg of lipid) of HARM suspension or with the same amount of HBS used as a control. Thirteen mice were used in the study. They were divided into 3 groups (1 group of three animals , and 2 groups of 5 animals). The rear flanks of the animals were shaved prior to receiving injections. The group 1 animals received a single 250 µl of the control, and were euthanized at 15 days. The group 2 and 3 animals received a single 250 µl control injection at a first site, and 250 µl injection at a second site. The group 2 animals were euthanized at 8 days, and the group 3 animals were euthanized at 15 days. The health of the animals was monitored daily.
Histology analysis was performed on tissue collected from the injection sites and preserved in 10% neutral buffered formalin solution. Cross sections from skin injection sites and surrounding tissue were processed by normal paraffin embedding and staining by Hematoxylin and Eosin.
The health of all 13 animals were normal throughout the study, and
there were no consistent changes in body weight. Histopathological
evaluation showed mild incidence of dermatitis, folliculitis and
perifolliculitis. However, these effects likely were the result of
trauma caused by shaving or injection, and not some negative reaction
to the injected material. The study demonstrates that there was no
gross toxicity associated with the tested samples.
The project is operating on schedule. Work is more advanced in
some areas, largely because of decisions not to pursue areas that do
not for the moment seem competitive with the work being pursued. We
have made a convincing demonstration that slow 0th order enzymatic hydrolysis of lipid tubules is
possible, even in the face of irregularities in the structure of the
lipid tubules used. We have found that almost every surfactant that
we have synthesized can be made into lipid tubules, helices, or
cochleate cylinders under appropriate conditions. We have found that
some combinations of tubule-forming lipids with different headgroups
appear to mix ideally, demonstrating the potential for mixing small
and large headgroup lipids for drug delivery. Studies on
detergent-driven dissolution suggest that tubules can be used for
rapid first-order drug delivery or vaccination via the intestines
following oral ingestion. We foresee both commercial interest in this
work from the drug delivery community as well as great basic research
interest in these novel enzymatic substrates.
1. Nakashima, N., S. Asakuma, J.M. Kim, and T. Kunitake, Helical superstructures are formed from chiral ammonium bilayers. Chem. Lett., 1984: p. 1709-1712.
2. Nakashima, N., S. Asakuma, and T. Kunitake, Optical microscopic study of helical superstructures of chiral bilayer membranes. J. Amer. Chem. Soc., 1985. 107: p. 510-512.
3. Yamada, K., H. Ihara, T. Ide, T. Fukumoto, and C. Hirayama, Formation of helical super structure from single-walled bilayers by amphiphiles with oligo-L-glutamic acid-head group. Chem. Lett., 1984. 10: p. 1713-1716.
4. Ihara, H., K. Yoshikai, M. Takafuji, C. Hirayama, and K. Yamada, Amphiphiles with polypeptide-head groups. 7. Relationship between formation of helical bilayer membranes and chemical structures of dialkyl amphiphiles with polypeptide-head groups. Kobunshi Ronbunshu, 1991. 48(5): p. 327-34.
5. Shimizu, T. and M. Hato, Self-assembling properties of synthetic peptidic lipids. Biochim. Biophys. Acta, 1993. 1147(1): p. 50-58.
6. Archibald, D.D. and S. Mann, Self-assembled microstructures from 1,2-ethanediol suspensions of pure and binary mixtures of neutral and acidic biological galactosylceramides. Chem. Phys. Lipids., 1994. 69(1): p. 51-64.
7. Nakashima, N., S. Asakuma, T. Kunitake, and H. Hotaini, Dynamic transformation of the morphology of dialkylammonium bilayer aggregates. Chem. Lett., 1984: p. 227-230.
8. Yager, P. and P.E. Schoen, Formation of tubules by a polymerizable surfactant. Mol. Cryst. Liq. Cryst., 1984. 106: p. 371-381.
9. Archibald, D.D., Structural studies of high aspect-ratio self-assembled lipid microstructures with the use of microscopy and FT-NIR-Raman spectroscopy, . 1990, University of Washington.
10. Goodman-Snitkoff, G., L.E. Eisele, E.P. Heimer, A.M. Felix, T.T. Andersen, T.R. Fuerst, and R.J. Mannino, Defining minimal requirements for antibody production to peptide antigens. Vaccine., 1990. 8(3): p. 257-62.
11. Gould-Fogerite, S., Y. Edghill-Smith, M. Kheiri, Z. Wang, K. Das, E. Feketeova, M. Canki, and R.J. Mannino, Lipid matrix-based subunit vaccines: a structure-function approach to oral and parental immunization. AIDS Res. Hum. Retroviruses, 1992. 10 Suppl 2: p. S99-103.
12. Mannino, R.J. and S. Gould-Fogerite, Lipid matrix-based vaccines for mucosal and systemic immunization. Pharm. Biotechnol., 1995. 6: p. 363-87.
13. Miller, M.D., S. Gould-Fogerite, L. Shen, R.M. Woods, S. Koenig, R.J. Mannino, and N.L. Letvin, Vaccination of Rhesus monkeys with synthetic peptide in a fusogenic proteoliposome elicits simian immunodeficiency virus- specific CD8+ cytotoxic T lymphocytes. J. Exp. Med., 1992. 176(6): p. 1739-44.
14. Kohler, E., M. Duberow-Drewe, J. Drewe, G. Riber, M.M. Loubatieres-Mariani, N. Maer, K. Gyr, and C. Beglinger, Absorption of an aqueous solution of a new synthetic somastatin analogue administered to man by gavage. Eur. J. Clin. Pharmacol., 1987. 33: p. 167-161.
15. Muranishi, S., M. Masahiro, M. Hashidzume, K. Yamada, S. Tajima, and Y. Kiso, Trials of lipid modification of peptide hormones for intestinal delivery. J. Controlled Release, 1992. 19: p. 179-188.
16. Gregoriadis, G., Liposome technology, ed. G. Gregoriadis. Vol. 1. 1984, Baco Raton, Fla.: CRC Press.
17. Gregoriadis, G., Immunological adjuvants: a role for liposomes. Immunol. Today, 1990. 11: p. 87-97.
18. Rogers, J.A., G.V. Betageri, and Y.W. Choi, Solubilization of liposomes by weak electrolyte drugs. I. Propranolol. Pharm Res, 1990. 7(9): p. 957-61.
19. Lichtenberg, D., Y. Zilberman, P. Greenzaid, and S. Zamir, Structural and kinetic studies on the solubilization of lecithin by sodium deoxycholate. Biochemistry, 1979. 18(16): p. 3517-3525.
20. Israelachvili, J.N., Intermolecular and Surface Forces. 1989 ed. 1985, London: Academic Press. 296.
21. Dennis, E.A., Micellization and solubilization of phospholipids by surfactants. Adv. Colloid Interface Sci., 1986. 26(2-4): p. 155-75.
22. Bakatselou, V., R.C. Oppenheim, and J.B. Dressman, Solubilization and wetting effects of bile salts on the dissolution of steroids. Pharm. Res., 1991. 8(12): p. 1461-1469.
23. Naylor, L.J., V. Bakatselou, and J. Dressman, Comparison of the mechanism of dissolution of hydrocortisone in simple and mixed micelle systems. Pharm. Res., 1993. 10(6): p. 865-870.
24. Luner, P.E., S.R. Babu, and G.W. Radebaugh, The effects of bile salts and lipids on the physicochemical behavior of gemfibrozil. Pharmaceutical Research, 1994. 11(12): p. 1755-1760.
25. Attwood, D. and A.T. Florence, Surfactant systems: their chemistry, pharmacy, and biology. 1983, London: Chapman and Hall.
26. Helenius, A. and K. Simons, Solubilization of membranes by detergents. Biochim. Biophys. Acta, 1975. 415: p. 29-79.
27. Jackson, M.L., C.F. Schmidt, D. Lichtenberg, B.J. Litman, and A.D. Albert, Solubilization of phosphatidylcholine bilayers by octyl glucoside. Biochemistry, 1982. 21: p. 4576-4582.
28. Chan, A.F., D.F. Evans, and E.L. Cussler, Explaining solubilization kinetics. AIChE, 1976. 22(6): p. 1006-1012.
29. Lichtenberg, D., R.J. Robson, and E.A. Dennis, Solubilization of phospholipids by detergents: structural and kinetic aspects. Biochim. Biophys. Acta, 1983. 737: p. 285-304.
30. Paternostre, M., O. Meyer, C. Grabielle-Madelmont, S. Lesieur, M. Ghanam, and M. Ollivon, Partition coefficient of a surfactant between aggregates and solution: application to the micelle-vesicle transition of egg phosphatidylcholine and octyl b-D-glucopyranoside. Biophys. J., 1995. 69: p. 2476-2488.
31. Tanford, C., Mixed micelles. 2nd ed. The hydrophobic effect: formation of micelles and biological membranes. 1980, New York: Wiley.
32. Maggio, B., T. Ariga, J.M. Sturtevant, and R.K. Yu, Thermotropic behavior of glycosphingolipids in aqueous dispersions. Biochem., 1985. 24: p. 1084-1092.
33. Haas, N.S. and G.G. Shipley, Structure and properties of N-palmitoleoylgalactosylsphingosine (cerebroside). Biochim. Biophys. Acta., 1995. 1240(2): p. 133-41.
34. Bohlen, P., S. Stein, K. Imai, and S. Udenfriend, A simplified protein assay with fluorescamine in samples containing interfering material. Anal. Biochem., 1974. 58(2): p. 559-62.
35. Marsh, D., CRC Handbook of Lipid Bilayers. 1 ed. 1990, Boca Raton, FL: CRC Press. 387.
36. Yager, P., A novel mechanism for the Na-K ATPase. J. Theor. Biol., 1977. 66: p. 1-11.
37. Yager, P. and P. Schoen, The formation of tubules by a polymerizable surfactant. Mol. Cryst. Liq. Cryst., 1984. 106: p. 371-381.
38. Yager, P., P.E. Schoen, C. Davies, R.R. Price, and A. Singh, Structure of lipid tubules formed from a polymerizable lecithin. Biophys. J., 1985. 48: p. 899-906.
39. Yager, P., R.R. Price, J.M. Schnur, P.E. Schoen, A. Singh, and D.G. Rhodes, The mechanism of formation of lipid tubules from liposomes. Chem. Phys. Lipids, 1988. 46: p. 171-179.
40. Ratna, B.R., S. Baral-Tosh, B. Kahn, J.M. Schnur, and A.S.
Rudolph, Effect of alcohol chain length on tubule formation in
1,2-bis(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine. Chem. Phys. Lipids,
1992. 63: p. 47-53
|
|
Return to <Yager's Home Page or to Tubule Project Page> |
|
 A
A
 B
B
 C
C
 A
A
 B
B
 A
A
 B
B

 I
I
 II
II
 III
III
 IV
IV
 V
V
 VI
VI
 Ia
Ia
 IIa
IIa
 Ib
Ib
 IIb
IIb
 Ic
Ic
 IIc
IIc
