|
Bioengineering Department, Box 355061, University of Washington, Seattle, WA 98195, USA |
 |
Self-Assembled Prodrug Tubules for Continuous Release
|
|
Bioengineering Department, Box 355061, University of Washington, Seattle, WA 98195, USA |
|
Delivering therapeutic drugs to the intended site of action at a controlled rate is an important goal of modern medicine. Lipid tubules are a recently discovered self-organizing system in which surfactants crystallize into tightly packed bilayers that spontaneously form cylinders less than 1 µm in diameter. Since the wide range of two-chain surfactants now known to form tubules includes lipidated polypeptides, it should be possible to synthesize tubule-forming lipids with polar headgroups that contain polypeptide and non-polypeptide drugs. These lipidated drugs might be active if the hydrophobic groups do not interfere with binding at their sites of action; alternatively, the tubule-forming drugs might function as prodrugs that become active when their hydrophobic moieties are cleaved. Because of the tight packing of the lipids in tubules, tubules should only dissolve or be enzymatically degraded from the tubule ends, resulting in a constant rate of tubule degradation until the tubules are consumed. Thus, these "prodrug tubules" could function as continuous release systems independent of any macroscopic encapsulation or delivery devices. The aims of the project are to determine in vitro whether tubules formed from model lipopeptides dissolve at constant rates, and to determine the structure of these tubules to establish principles for design of lipopeptides and other prodrugs of therapeutic value. Primary techniques to be employed are organic synthesis, calorimetry, optical and electron microscopies, and Raman and fluorescence spectroscopies.
Delivering therapeutic drugs to the intended site of action at a controlled rate is an important goal of modern medicine. Recent work has centered on fabrication of macroscopic devices that allow continuous release of some drugs under limited circumstances. We present a novel approach to continuous release in which neither macroscopic matrix nor pump is required, and the "device fabrication" is accomplished through molecular-level self-assembly. The long-term objective of the work is to develop a device-free method by which many drugs can be released into the body in a continuous manner (0th-order kinetics) through association with self-assembling tubular microstructures.
Lipid tubules are a recently discovered self-organizing system in which lipids crystallize into tightly packed bilayers that spontaneously form hollow cylinders less than 1 µm in diameter. Because of the tight packing of the surfactants in tubules, the microstructures should dissolve (or be enzymatically degraded) from their ends only. Since the size of the end (the only available surface area for removal of drug) is constant until the tubule is annihilated, a population of tubules of uniform length will release surfactant at a constant rate. Since the wide range of two-chain surfactants now known to form tubules includes lipidated polypeptides, and such lipidated polypeptides are composed of innocuous nontoxic biomolecules, it should be possible to synthesize tubule-forming lipids with polar headgroups that contain polypeptide and non-polypeptide drugs. These lipidated drugs might be active themselves if the hydrophobic groups do not interfere with binding at their sites of action; alternatively, the tubule-forming drugs might function as prodrugs that become active when their hydrophobic moieties are cleaved. The aforementioned tight packing of the lipids will be transferred to the drug headgroups. Such structures will either dissolve spontaneously at a rate controlled by structure-determined solubility of the lipidated prodrug molecules, or they might release drug by enzymatic degradation of the microstructures from their ends. These "prodrug tubules" could function as continuous release systems independent of any macroscopic encapsulation or delivery devices.
Tubule-based continuous release would probably have first applications in topical administration of drugs, antibacterials and antifungals, release of antitumor drugs/peptides by injections of tubules into or near tumors, antibiotic and growth factor delivery for wound dressings, long-acting vaccines, delivery of peptide and non-peptide drugs from subcutaneous sites, and in vitro use for tissue/cell culture factors.
The short-term objective of this proposal is to perform in vitro experiments that prove or disprove the following 3 hypotheses:
A homogeneous population of lipid tubules will dissolve (or be enzymatically degraded) in such a manner that the rate of release of the constituent molecules (or parts thereof) will be constant until the microstructures are consumed
Ligation to an appropriate hydrophobic anchoring moiety will allow both model water-soluble molecules and clinically significant drugs and prodrugs to self-associate into tubular microstructures. Of particular interest are bioactive polypeptides.
Tubule-associated drugs or prodrugs will continuously release drugs in simulations of drug delivery environments, either through dissolution of the molecules from tubule ends or through enzymatic cleavage from the tubules.
The Gelb and Yager groups provide complementary expertise in organic synthesis, enzymatic degradation of lipid bilayers and the self-assembly of lipids into tubules. Primary techniques to be employed are organic synthesis, calorimetry, optical and electron microscopies, and Raman spectroscopy. The work to be accomplished will provide the basis for exploitation of this new route of continuous release of drugs by establishing principles for design of tubule-forming lipopeptides and other prodrugs of therapeutic value.
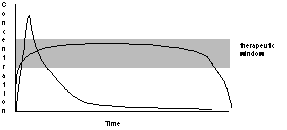
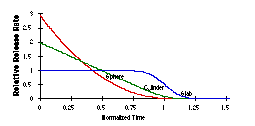
The two most important current issues in drug delivery are spatial and temporal: targeting to limit exposure to the desired site of action, and controlled delivery. Continuous release of drugs, preferably at the site at which they are most effective, is vastly preferable to periodically administering bolus doses to the entire organism. The administration of therapeutic drugs in bolus form results in a spike of drug concentration, followed by an exponential decay to baseline as in Figure 1. The decay in concentration can be caused by metabolism, excretion, or uptake by non-target organ systems, all of which result in there being less of the drug available to act at the intended site. Many drugs are toxic at high concentrations, so in defining a "therapeutic window" a compromise is made between using enough drug to effect a cure and minimizing the time at which the drug is at toxic concentrations. The "therapeutic window" is often narrow; even if the drug is not toxic the cost of treatment can be reduced if only as much drug is administered as is needed. In many cases one would either like to greatly prolong the time of release of the drug, reduce transient toxic levels of a drug at the beginning of administration, reduce the number of doses that must be administered while not exceeding a toxic threshold, or all of these. The ideal curve might look more like a square wave, rather than a capacitive transient.
|
|
Figure 1. Comparison of the in vivo concentration of a drug administered in a bolus dose (spike with exponential decay) with the time course administered "continuously". Above the therapeutic window the toxicity of the drug outweighs its benefits. |
At present there are several approaches to controlled or continuous drug delivery, some of which are still in the research phases, and some of which have been very successfully used in commercial products for some time [2]. The existence of many recent reviews of these delivery systems attests to the feverish rate of research in this field [3-8]. The delivery approaches range from external delivery systems such as external mechanical pumps and osmotic patches, to internal osmotic pumps, and implantable or ingestable polymeric structures that may include erodable hydrogels. With a pump, continuous release can be set by the design of the pump or control of the motor. With polymeric structures the rate of delivery can be controlled by the shape and permeability/erodability of the polymer. Some of these approaches work well for some classes of drugs, and are inapplicable to others; the chemically labile nature of peptide drugs results in their incompatibility with many polymeric delivery systems. Those polymers in which they can be immobilized [2] have yet to be approved for general use.
The explosion in our awareness of the importance of peptides as chemical signals has resulted in great interest in the use of peptides as drugs, but their use is fraught with problems, including vulnerability to premature proteolysis in vivo, the inability of hydrophilic peptides to cross biological membranes, skin, or the blood-brain barrier, the need for a targeting delivery system. They are, for these reasons, among the most difficult types of chemicals to use as drugs.
The common feature of all these existing delivery systems is that they involve the use of control of diffusion or effusion by a macroscopic mechanical object. If the only method of continuous delivery of a drug is continuous infusion with an i.v. line (as it is for some chemotherapeutic drugs), the cost is high and the patient's movement is restricted. Use of implanted catheters and pumps is an expensive solution, the considerable risk of which is only balanced by the importance of continuous delivery of the drug in question. Dermal patches are very simple and relatively noninvasive, but have been effective only for a few drugs that are relatively permeant through the skin. The use of implantable macroscopic devices for drug delivery restricts the site of delivery to one that can accommodate the object. The Norplant contraceptive device, effective though it is, requires a large insertion site and must be surgically recovered after use. Langer's use of erodable drug-laden polymer disks in post-surgical treatment of brain cancer is laudable [2], but would it not be better if there were a general method of continuously delivering the antitumor drugs to tumors before (or without) major surgery?
In summary, current research into continuous drug delivery does not suggest adequate answers to many drug delivery problems. What if one could have the advantages of prolonged steady delivery without the macroscopic size of the drug delivery devices currently under study?
It is now well understood that encapsulation of drugs in liposomes can be of great use in some circumstances [9], such as in delivery of drugs to skin [10]. Liposomes may also have a role in gel-based drug delivery systems [11]. A separate issues is covalently modifying hydrophilic drugs with hydrophobic moieties, which can greatly enhance drug utility by a) allowing them to cross cell membranes, epidermis and the blood-brain barrier, b) by facilitating in vivo targeting (by association with targeting liposomes, or restricting their movement to particular sites in the body), and c) (on which we will focus) by changing the kinetics at which the drugs are delivered. Several recent lines of work are exploring the effect of attaching hydrophobic moieties to peptides and other soluble drugs. Modification of some short peptides into "lipopeptides" by addition of 2 to 3 palmitoyl chains appears to turn the peptides into self-adjuvants [12], enhancing their ability to elicit an immune response. After addition of an N-terminal myristoyl group, one membrane-impermeable nonapeptide has been shown to cross lipid bilayers and to become an effective enzyme inhibitor [13]; the presence of the hydrophobic group did not adversely affect activity. Addition of a cleavable hydrophobic (steroid) carrier moiety has also been shown to allow a peptide to cross the blood-brain barrier [14]; cleavage of the hydrophobic group allowed intracellular generation of the unmodified active peptide.
How a drug is distributed can be controlled not only by its hydrophobicity, but also by the microstructures into which it self-assembles. Once a peptide is modified by hydrophobic groups, it becomes a surfactant, and depending on the relative sizes and shapes of the polar "head group" and the hydrophobic "tail", surfactants can take on many forms in water [15]. Self-association of such lipopeptides above their CMC (critical micelle concentration) will produce either micelles or bilayers, depending on the ratio of the projected areas of the polar and nonpolar moieties. Individual molecules may be able to pass through the blood brain barrier, whereas liposomes may be retained within the blood stream. Phosphatidylglycerols have been modified with a wide range of peptide and non-peptide drugs [16] (in particular AZT [17-25]) with the assumption that they would self-assemble into liposomes, and would, after injection into the bloodstream, be trapped by macrophages in the reticuloendothelial system. Beyond the general assumption that liposomes would be formed (with a few exceptions such as one study on model polypeptides [26]) how hydrophobically modified drugs self-associate, and how the self-association affects the conformation of the drugs themselves, is largely unknown.
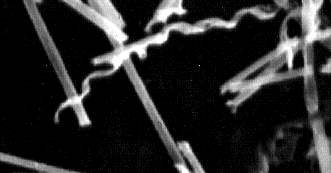
In 1983, polymerizable diacetylenic phosphatidylcholines such as 1,2-di-(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine (DC8,9PC) (Figure 4C, page 32) were discovered by Yager and Schoen to form novel hollow tubular microstructures (Figures 2,3) [27-30]. Diacetylenic lipid tubules are straight, rigid, about 0.75 µm in diameter, and can be made to range in length from a few µm to nearly 1 mm, depending on formation conditions. Further, the walls of the tubules may be as thin as a single bilayer, so a good model is a hollow straw. The lumen is generally open, allowing free access by diffusion from the ends of the microstructures.
|
|
|
|
Figure 2. Phase contrast optical micrograph of diacetylenic lipid tubules in water [31]. Quantitative conversion of diacetylenic phosphatidylcholine DC8,9PC by cooling of liposomes is possible [29], although reproducibility is better if they are formed (as in the figure) by precipitation from a solution of the lipid in alcohol by addition of water, [32]. |
Figure 3. Scanning electron micrograph of (metallized) DC8,9PC tubules formed by solvent precipitation. Diameters ~0.75 µm. Under some precipitation conditions helical ribbons are seen as a minority population, but in most cases the helical ribbons fuse to continuous cylinders. If not coated with metal, the surface tension of water flattens tubules when they are dried in vacuo. |
The basic subunit of the tubule is a helical ribbon of lipid bilayer (Figure 8), and, in some cases, open helical structures of the same diameter can be seen (Figure 3). Unlike liposomal bilayers, the tubule wall is a single crystal, which has been demonstrated by low-dose electron diffraction; a single diffraction pattern is seen along the entire length of the microstructure (Price and Yager, unpublished results). Tubules and helical microstructures convert reversibly into sealed liposomes on heating through Tm, the temperature at which the gel phase melts into the liquid crystalline phase. All symmetrically disubstituted diacetylenic PCs synthesized to date have been found to form tubules and helices under appropriate formation conditions.
For a brief time it was believed that zwitterionic diacetylenic lecithins such as DC8,9PC (Figure 4C) were the only amphiphiles that produced tubules and helices. Markowitz and Singh have shown that phospholipase D can be used to change the headgroup of DC8,9PC from phosphocholine to phosphoethanol, giving the lipid a net negative charge at pH 7. At pH 5.6 in distilled water, tubules do not form, but in the presence of 100 mM monovalent salt or millimolar divalents, tubules form and are stable [33]. Members of a family of synthetic single chain amphiphiles (N-octyl-aldonamides) were shown by Führhop to form narrower rods, whiskers, helices, and cochleate cylinders with axial ratios as high as 104 [34, 35]. Kunitake's group demonstrated that a positively charged chiral amphiphile based on glutamate (Figures 4A, 5A) [36, 37] forms structures similar to those formed by DC8,9PC. On cooling of liposomes of this lipid through Tm, quantitative conversion to helices occurs. Over a week these helices convert to intact tubules with homogeneous diameters nearly identical to those formed by the diacetylenic PCs. Helices and tubules of much smaller diameters (~300Å) were found by Yamada et al. to form from related synthetic two-chain amphiphiles with oligopeptides (such as 12-14-mers of glutamic and aspartic acid) as hydrophilic headgroups [38, 39] (Figure 4B, 5B). Recent work by Shimizu and Hato on similar lipids with polypeptide headgroups including (Pro)3 tripeptide produced similar tubules and helices (Figure 5C) [40]. The very similar behavior of the Yamada, Shimizu and Kunitake lipids implies that ordered hydrophobic regions are largely responsible for the crystalline microstructures, and that a wide variety of peptides could be used as polar headgroups. Later studies by the Yamada group ascertained that both positive, negative and neutral amino acids could be incorporated into block copolymers as headgroups for glutamate-based lipopeptides; but that fully charging the headgroups prevented tubule and helix formation [41]. This is presumably because charging the polypeptide side chains increases the headgroup excluded volume to the point that close packing of the hydrocarbon chains is no longer possible in a planar bilayer. Further, there was evidence that the secondary structure of the polypeptide varied with the nature of the microstructure [39], and that b-sheet formed between headgroup polypeptides. However, there is little data to support the contention that formation of a particular type of secondary structure is required for tubule formation.
|
|
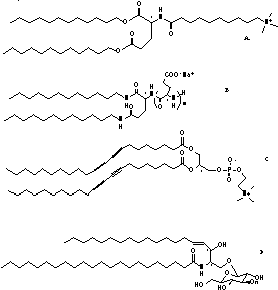
Figure 4. Structures of representative tubule-forming surfactants: A. Glutamate-based amphiphile from
Nakashima in Kunitake's laboratory [36, 37, 42]. B. Polyglutamate-based amphiphile
explored by Yamada's group [38]. C. Phosphatidylcholine with
tricosadiynoyl fatty acyl chains (DC8,9PC, studied extensively
by Yager et al.) [27]. This lipid is one of a class of
"diacetylenic phospholipids". D. The fraction of bovine brain cerebroside in which the fatty acyl chain is without a 2-hydroxyl group (known as NFA-cer) [43]. | |
|
|
| |
|
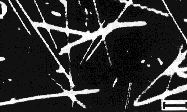
A. Tubules and helices formed by the glutamate-based amphiphile shown in Figure 4A [37]. Darkfield optical micrograph (poorly reproduced), scale bar = 10 µm. |
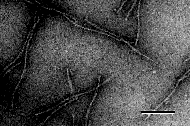
B. Small tubule-like microstructures formed at pH 4 by the glutamine-based amphiphile in Figure 4B [39]. Negative stain TEM, scale bar 0.1 µm. | |
|
|

| |
|
C. Tubules and helices formed by a glutamate-based amphiphile with a headgroup consisting of a (Pro)3 tripeptide. Darkfield optical micrograph, scale bar = 10 µm. [40] |
D. TEM of Small tubule-like microstructures formed by a glutamine-based amphiphile with positively charged aromatic headgroup [41] | |
Figure 5. Microstructures formed by synthetic amphiphiles. Those in A and C are large structures very similar to those formed by diacetylenic phosphatidylcholines, whereas those in B and C are substantially narrower and are perhaps more similar to those formed by NFA cerebrosides.
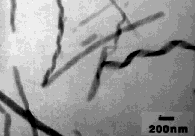
At first, only synthetic amphiphiles were known to form these structures. (Although the Kunitake and Yamada lipopeptides are formed from natural compounds, the whole surfactants are not known to be formed in nature.) However, the Yager group recently found that helical and tubular structures (Figure 6), as well as rod-like cochleate cylinders (see schematic in Figure 8), can be formed quantitatively from the n-fatty acyl (Figure 4D) and a-hydroxy fatty acyl fractions of bovine brain galactocerebrosides, designated NFA-cer and HFA-cer, respectively [44]. Both NFA-cer and HFA-cer have heterogeneous mixtures of fatty acyl chains, some of them unsaturated [45], and yet they still form crystalline microstructures at room temperature. Cerebroside microstructures are identical to those found in human tissue in several lipid storage diseases [46-48]. Evidently a good deal of variability in the headgroup is possible, as a large fraction of charged sulfated cerebrosides can be added to NFA-cer without preventing tubule formation [49, 50]. Even D-erythro-sphingosine (which lacks both the sugar and a fatty acid of NFA-cer), forms 50-nm diameter flexible fibers, rather than liposomes [51]. Tubular and helical structures have now been observed in samples of aged suspensions of saturated-chain phosphatidylcholines [52], and as transient intermediates in the crystallization of cholesterol from mixed micellar suspensions (Figure 7) [53, 54].
|

|
|
Figure 6. TEM of small tubules and helices formed by NFA galactocerebrosides from bovine brain [44]. The edges of the helices can fuse to form straight hollow tubules [27, 29]. |
Figure 7. Optical micrograph of large tubules and helices of pure cholesterol formed in the presence of bile [54]. |
|
|

|
|
|
|
|
|

|
|
|
|
Figure 8. Comparison of known non-liposomal microstructures of bilayer-forming amphiphiles. The formation of twisted ribbons, helices, and tubules is driven by cholesteric interactions between adjacent chiral amphiphiles: cochleate cylinders need not have similar interactions, but require strong bilayer-bilayer interactions
Several theories of why tubules form have been published recently [55-62], and while many of them are successful in describing the morphology of tubules and their relationship to twisted helical strips, none of them (including that from the Yager group) deals with the molecular details within the crystalline bilayer. The empirical result is that tightly packed two-chain surfactants are found to self-assemble into at least the four thermodynamically stable non-liposomal microstructures found in Figure 8. Whether they form one structure or another depends on details of the molecular packing, but from the fact that so many different lipids form tubules, clearly no single specific molecular structure is required. We believe that the fundamental interaction between surfactants in tubules, twisted bilayer ribbons and helical ribbons is one in which there is a cholesteric interaction (as in cholesteric liquid crystals) in the plane of the bilayer. The recent discovery of the formation of tubules and helices formed from cholesterol itself strengthens this argument [53, 54]. Our view is that a surfactant can form helices and/or tubules if it is chiral, forms bilayers, crystallizes in that bilayer with tight inter-chain interactions, and if it does not have very strong inter-bilayer interactions[31, 57]. If inter-bilayer forces dominate, the result is a cochleate cylinder.
Although lipid tubules continue to be considered technologically promising because of their unique combination of straightness, rigidity and small size, there have been no commercialized uses for them to date. Lipid tubules have been "decorated" with inorganic materials, including metals [63, 64] and salts [65]. Metallization, in particular, has allowed them to be tested for several applications, including hollow metal containers for antibiotics in antifouling marine paint, and for field emission electrodes [66]. Recent publications have focused public attention on selective decoration of tubules with metal oxides by varying the relative concentrations of three cerebrosides with different headgroups [49, 67-69]; this "tubule design" is an extension of work in the Yager laboratory on the cerebroside system [43, 44]. Some preliminary work has also been undertaken to use the lumen of diacetylenic lipid tubules as a reservoir for the encapsulation of drugs for delivery in wound dressings [66, 70, 71]. Optical and electron microscopy by Rudolph et al. also showed adherence of monocytes and other human cell lines to lipid tubules with and without an admixture of gangliosides [72]. However, the drug delivery approach suggested below is distinctly different, and potentially much more widely applicable.
Lipid tubules may present a solution to many continuous drug delivery problems because, as is well known, the geometry of drug particles affects the kinetics of drug release. The key fact is that tubules are single crystals, and like all crystals, can only dissolve from their surfaces. Unlike most 3-D crystals (or amorphous materials, for that matter), the free surface area for loss of surfactant to the solution exists only at the ends of the tubules. If a tubule dissolves or is broken down only from its ends, the rate of appearance of dissolved surfactant or surfactant breakdown products must remain constant until the number of tubules (and ends) declines. If the rate of drug release to the tissue is limited by the rate of release from the ends of rodlike microstructures (Figure 9), the drug release rate will be constant (0th-order), as opposed to the more conventional first-order kinetics found with a wide range of other geometries.
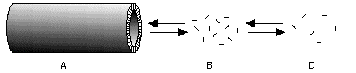
Figure 9. A crystalline tubule (A) of a tubule-forming lipopeptide drug can dissolve only by loss of individual molecules from its ends. The tubule ends will tend toward equilibrium with a volume in which the lipopeptide is at or above its CMC (B); this volume will, in turn, exchange with the site of delivery © by diffusion, etc.
There are two ways in which tubules could be used to produce continuous release of drugs, as described below. They differ in that in one mechanism the release of drugs in dependent only on the dissolution of the drug from the ends of tubules, but in the other method the drug is released from the tubule under the influence of an enzyme-catalyzed hydrolysis reaction, in which the enzyme is presumably supplied by the environment in which the tubules are localized.
In the first scenario as shown in Figure 10, the tubule-forming molecule must consist of a tubule-forming surfactant with a drug covalently attached to its headgroup. The surfactant would either be a lipidated drug if it were active in its intact form, or a lipidated prodrug if it were activated by enzymatic or nonenzymatic cleavage after entering the target cell. The constant rate of dissolution of the tubules would be controlled largely by the solubility of the lipidated drug in the surrounding medium. The greater the ratio of head group area to hydrocarbon chain surface area, the more rapid will be the dissolution and delivery.
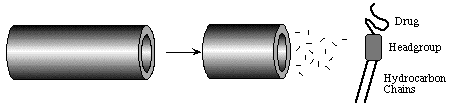
Figure 10. Schematic representation of the dissolution of monomeric lipidated drugs from the edge of a tubule bilayer. As the bilayer is crystalline and nearly defect-free, dissolution is only possible at tubule ends.
In the second approach, the drug moiety is attached to the tubule-forming surfactant via a cleavable spacer group. Spacers might be a polypeptide with a sequence known to be susceptible to attack by a protease at the site of use (Figure 11), or a functional group of limited stability against cleavage when exposed to the solution at the site of use. Packing of the drugs at the tubule surface would be tight enough to prevent access by a protease. Only at the disordered tubule ends would there be access for enzyme (see Figure 12), so the release of the drug would be controlled by the constant number of intact tethers exposed at the advancing front of tether cleavage.

Figure 11. Representation of the second form of tubule-based drug delivery. The coating of the outer (and inner) surfaces of the tubule by a close-packed layer of drug molecules is represented by the dot-covered surface of the tubules. Enzymes capable of cleaving the linkage of the drug to the tubule-forming surfactant can gain access to the lipid-drug tether only at the disordered ends of the tubules (at left), and over time strip the tubules of drug from the ends inward at a constant rate.

Figure 12. Schematic of a 5x8 monolayer array of lipids at the edge of a tubule (at left) being consumed by enzymatic hydrolysis. At point A (representing the "virgin" tubule surface) the packing of headgroups is too tight to allow access by enzymes to either the cleavable tether or the tubule-forming lipid. Attack of the tether is limited, therefore, to the advancing front of enzymatic attack (B). While the remaining lipid will have a low aqueous solubility, the extreme edge of the remaining bilayer © will be accessible to subsequent lipolytic enzymatic attack, removing drug-free lipids from the tubule, and ultimately dissolving it.
While this approach seems more complex, it offers the exciting possibility of employing a single standard tubule-forming surfactant and linker group for coupling to a wide range of water-soluble molecules, including biomolecules such as polypeptides and nucleic acids.
We envision the use of tubules as extracellular vehicles for the delivery of various types of drugs that are initially part of the tubule microstructure. The tubules will be composed of drugs covalently linked to natural biodegradable moieties such as ceramides, phosphatidylcholines, amino acids and fatty acids; the structural components should eventually be completely metabolized into nontoxic products* . While the physical size of tubules will prevent them from being used in all applications, a suspension of tubules should be usable in all circumstances for which macroscopic polymeric drug delivery systems are currently contemplated. This method of controlled release avoids pumps or incorporation of drug into a macroscopic rigid matrix of a particular shape. The small diameter of tubules allows them to be placed into cavities in the body using a needle or catheter, whereas their great length will immobilize them after injection. For example, a wide range of tubule-based antitumor drugs could be injected into tumors through small needles, perhaps avoiding the need for major surgery in some cases.
Another promising use of tubule-based drug delivery systems in the short run would be in controlled release in topical or subcutaneous applications in which the great length of some of the microstructures could immobilize them without a rigid polymeric matrix. Topical use, in particular, could be tested in the near term, as ceramides, at least, are approved for use in cosmetics. They might also find use in mucosal and oral delivery. The tight packing of the lipid molecules in the tubule could afford protection of certain drugs such as peptides from the premature enzymatic hydrolysis that now plagues peptide delivery systems. While there are often ample concentrations of proteolytic and lipolytic enzymes present in the interstitial fluid in vivo , these enzymes are often inhibited to prevent uncontrolled cell damage [73-75]. Whether normal levels of such enzymes in interstitial fluid would be adequate to hydrolyze lipopeptide tubules remains to be determined. To ensure that enzymatic release of drug from tubules will occur in an extracorporeal site such as in topical applications or in vitro, tubules could be co-suspended with hydrolytic enzymes.
While there is nothing inherently antigenic about a lipid tubule, subcutaneous injection of some drug-coated tubules may induce an inflammatory response, as demonstrated by the adherence of some cells to DC8,9PC tubules [72]. The cellular environment in the presence of such a response will provide ample proteolytic enzymes to cleave prodrugs from the tubule surfaces, which could be an advantage. Some vaccination protocols require repeated dosing with vaccines because a single bolus dose does not raise an adequate immune response. Tubules placed in subcutaneous sites could act as long-acting vaccines that deliver antigen long enough to create a strong immune response.
Because the rate of degradation of lipidated drugs will be quite different depending on whether the surfactants are in the form of tubules or liposomes, it is possible that raising the local temperature above Tm, which will convert the tubules to liposomes, could provide a method of greatly increasing the delivery rate from implanted microstructures on demand.
Even if it proves that in vivo use of tubules is restricted for some reason, the continuous release using tubules could still be important in such in vitro applications where delivery of some chemical is required over a long period at a constant rate. A biotechnologically important example would be the delivery of growth factors or antibiotics to cells being cultured in containers too small to merit continuous infusion of such factors.
As shown below, the rate of drug release will only be constant to the extent that the tubule population is homogeneous in length. While is it possible to form tubules with unimodal length distributions by the crystallization methods we have developed [32], there is always a distribution of lengths about the mean. We have demonstrated that storage often results in the smaller tubules converting to longer ones, and that it is possible to remove the extremes of the length distribution using filtration and sedimentation.
Larger diameter tubules (~ 1 µm diameter) are unlikely to be safe for injection into the circulatory system because of possible clogging of capillaries. All other internal and external sites of drug delivery are possible, however. Because so much of the mass of the tubule is in the wall means that there is a large "wasted" central lumen in the tubules, reducing the possible drug loading. Multi-bilayer tubules or cochleate cylinders may be better suited to circumstances where high drug loading is necessary. However, most drugs are potent enough that the hollow tubule lumen will not pose a problem. While small molecules will escape rapidly from this cavity, larger ones of therapeutic interest could be encapsulated for release. Smaller and more flexible tubules like those shown in Figure 5B & D (~<0.1 µm diameter) have less wasted space and may also be small enough to pass through the capillary beds.
Each in vivo site will have a unique set of hydrolysis conditions (pH, enzymes, etc.), so the delivery system (cleavable linkers, etc.) will have to be tailored for the site. There is a small chance that peptide- or drug-coated tubules could be immunogenic, depending on how they are processed by the immune system. While this would be an advantage for use in vaccines, it could restrict the use of drug-delivery tubules use to immunologically privileged sites.
The amphiphile N,N'-dihexadecyl -L-glutamine amide (compound 1 in Figure 19) is the core of the Yamada lipopeptide (Figure 4B). This lipid was not extensively characterized in the literature, despite its role as the hydrophobic core of several more elaborate lipids. We have now synthesized it several times following the method described by Yamada et al. [38, 76]. Hexadecylamine was coupled to both of the free carboxyl groups of N-carbobenzoxy-L-glutamic acid with diethyl cyanophosphonate in the presence of triethylamine to form amide linkages. Cleavage of the protecting carbobenzoxy group was accomplished by hydrogenation with 10% Pd on activated carbon. The physical properties of compound 1 are now under study. Studies include differential scanning calorimetry, and optical and electron microscopy of its aqueous dispersions to determine if it forms tubular microstructures.
If our end-dominated model of the dissolution and lysis of tubules is to be proven correct, experimental kinetic data must be differentiable from data to be expected for different models. We compared theoretical dissolution rates of three structures-a solid sphere, an infinitely long solid cylinder, and a slab (which models the kinetics for dissolution at tubule ends).
|
|
|
|
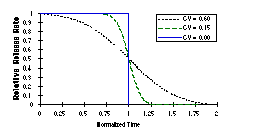
Figure 13. Model of the effect of polydispersity of tubule length on the rate of dissolution (and delivery) of drug as tubules dissolve from their ends. Dissolution time is normalized so a tubule with the mean length dissolves completely by time 1.0. CV stands for 'coefficient of variance' of a Gaussian length distribution. Graphs are normalized so that the total mass released is the same. |
Figure 14. Comparison of kinetics of dissolution of spheres, infinitely long solid cylinders (no diffusion from the ends), and flat slabs (which model the dissolution from the ends of tubules). The CV for each plot in this figure is 0.15. Graphs are normalized so that the total mass released is the same. |
In all cases the dissolution rate is proportional to the exposed surface. The three are drastically different when considering one particle or a homogeneous population of particles. However, heterogeneity in particle size softens the distinction between the models. For example, in Figure 13 is shown the effect of broadening the coefficient of variance (distribution of tubule lengths or slab thicknesses). However, as shown in Figure 14, broadening the distribution still allows the nature of the dissolution to be distinguished by the number and position of inflection points in the delivery rate curve. It should still be possible to determine if our model of end-dominated dissolution and lysis is correct from delivery rates alone.
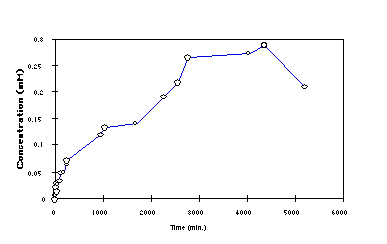
A proof-of-principle experiment was performed on DC8,9PC, which is commercially available (Avanti Polar Lipids, Birmingham AL). Our hope was to prove that tubules could be enzymatically cleaved (and release fatty acid) at a constant rate. The enzyme phospholipase A2 (PLA2) is known to hydrolyze the fatty acid at the 2 position of the glycerol backbone of phospholipids. It is also known that PLA2 only binds tightly to bilayers in the presence of negatively charged lipids such as fatty acids; once some hydrolysis has occurred, the proportion of membrane-bound enzyme increases. After confirming that PLA2 can hydrolyze the well-studied dipalmitoyl phosphatidylcholine (DPPC) below its Tm, we set out to determine whether the enzyme can work on DC8,9PC below its Tm in tubule form. To monitor the concentration of free diacetylenic fatty acid product, the intestinal fatty acid binding protein ADIFAB (Molecular Probes, Eugene, OR) was used. ADIFAB, which is labeled with acrylodan, undergoes a change in fluorescence emission wavelength when it binds fatty acids. We found that this change accurately monitored the concentration of free diacetylenic fatty acid.
Optical microscopy showed that, at least in the presence of 10 mM Ca++ required for PLA2 activity, the only visible change in a tubule suspensions after a day of hydrolysis was that the tubules began to stick to each other. We attribute this to "crosslinking" of tubule surfaces by the action of Ca++ on of the negatively charged fatty acids produced by the hydrolysis. We expected an excess of the detergent Triton S-100 (TX100) to disperse tubules immediately, but even 10-fold and 100-fold molar excess (far above the CMC for this detergent) had no apparent effect on the tubules over several hours. Over a period of a week the 100-fold excess did dissolve nearly all of the tubules, but little effect was apparent with a 10-fold excess of TX100. On the other hand, heating the tubules above Tm in even a 10-fold molar excess of TX100 instantly and irreversibly clarified the suspension by converting the tubules to small mixed micelles. This confirms earlier suggestions that the surface of the crystalline structure of the DC8,9PC tubules is completely impervious to penetration within the surface of the tubule wall [77]. It is probably only at the ends that detergent dissolution is possible below Tm. We will investigate the kinetics of the TX100 dissolution within the next few months.
|
|
Figure 15. The total concentration of diacetylenic fatty acid vs. time as produced by the action of PLA2 on a suspension of DC8,9PC tubules, as determined using the ADIFAB binding assay. The buffer contained 50 mM Tris-HCl (pH 8.0), 10 mM CaCl2, 0.05% sodium azide, 0.5 mM DC8,9PC and 4 units Naja naja venom PLA2. The 0.25 mM concentration achieved by 3000 minutes represents ~50% completion of the hydrolysis reaction. Note that an initial phase of rapid hydrolysis is followed by a long period of relatively constant enzymatic production of fatty acid. |
The results shown in Figure 15 indicate that after an initial spike of rapid hydrolysis (perhaps caused by the presence of small tubule fragments in the sample) PLA2 -catalyzed hydrolysis of tubule DC8,9PC proceeded with 0th order kinetics for a period of nearly three days until approximately 50% of the lipid was hydrolyzed. The experiment was terminated in order to finish writing this grant application. The reason for deviation of the last point is unknown, and under investigation; the decrease may have been due to air oxidation of the diacetylenic fatty acid, or to the extreme sensitivity of crystallites of pure fatty acid to polymerization, which would render them insoluble and therefore undetectable by ADIFAB. Tubules were still present in the sample at the end of the experiment. We will repeat this experiment using a pH stat to measure fatty acid production in the near future. A model of what appears to happen is shown in Figure 16, which is similar to the action of proteolytic enzymes on the crosslinker in Figure 12. Note that in liposomes the products of PLA2 hydrolysis remain in the bilayer unless albumin is present to bind fatty acid [78].
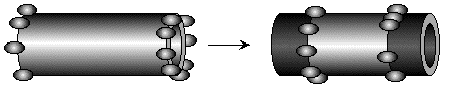
Figure 16. PLA2, as an example of a lipolytic enzyme with a well-studied mechanism, appears to attack lipid tubules from the ends only, and then proceeds inward along the tubules, converting phospholipid to lysolipid and fatty acid. It is still not clear whether these particular hydrolysis products remain in tubule form or diffuse away.
To determine whether there was some intrinsic incompatibility between cells and tubules, washed DC8,9PC tubules formed as mentioned above were dispersed into a culture of rat basophilic leukemia cells (RBL-2H3) and incubated for 2-3 days. Normal tubules were observed to be present at the end of the test period, and their number did not change, and the cells continued to grow at their normal rate.
9. Literature Cited.
|
1. |
Reach, G. and G.S. Wilson, Can continuous glucose monitoring be used for the treatment of diabetes. Analytical Chemistry, 1992. 64(6): p. 381A-386A. |
|
2. |
Langer, R., New methods of drug delivery. Science, 1990. 249(4976): p. 1527-33. |
|
3. |
Prevost, G., et al., Therapeutic use and perspectives of synthetic peptides in oncology. Acta. Oncol., 1993. 32(2): p. 209-15. |
|
4. |
Oliyai, R. and V.J. Stella, Prodrugs of peptides and proteins for improved formulation and delivery. Annu. Rev. Pharmacol. Toxicol., 1993. 33: p. 521-44. |
|
5. |
Harris, D. and J.R. Robinson, Drug delivery via the mucous membranes of the oral cavity. J. Pharm. Sci., 1992. 81(1): p. 1-10. |
|
6. |
Pardridge, W.M., Recent developments in peptide drug delivery to the brain. Pharmacol. Toxicol., 1992. 71(1): p. 3-10. |
|
7. |
Pardridge, W.M., Opioid peptide drug development: transport of opioid chimeric peptides through the blood-brain barrier. NIDA Res. Monogr., 1992. 120: p. 153-68. |
|
8. |
Muranishi, S., Absorption enhancers. Crit. Rev. Ther. Drug. Carrier Syst., 1990. 7(1): p. 1-33. |
|
9. |
Ostro, M.J., ed. Liposomes: From Biophysics to Therapeutics. . 1987, Marcel Dekker, Inc.: New York. 393. |
|
10. |
Moghimi, S.M. and H.M. Patel, Current progress and future prospects of liposomes in dermal drug delivery. J. Microencapsul., 1993. 10(2): p. 155-62. |
|
11. |
Wu, X.S., A.S. Hoffman, and P. Yager, Conjugation of phosphatidylethanolamine to poly(N-isopropylacrylamide) for potential use in liposomal drug delivery systems. Polymer, 1992. 33(21): p. 4659-4662. |
|
12. |
Metzger, J., et al., Lipopeptides containing 2-(palmitoylamino)-6,7-bis(palmitoyloxy)heptanoic acid: synthesis, stereospecific stimulation of B-lymphocytes and macrophages, and adjuvanticity in vivo and in vitro. J. Med. Chem., 1991. 34: p. 1969-1974. |
|
13. |
Eichholtz, T., et al., A myrisoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J. Biol. Chem., 1993. 268(3): p. 1982-1986. |
|
14. |
Bodor, N., et al., A strategy for delivering peptides into the central nervous system by sequential metabolism. Science, 1992. 257(5077): p. 1698-700. |
|
15. |
Israelachvili, J.N., Intermolecular and Surface Forces. 1989 ed. 1985, London: Academic Press. 296. |
|
16. |
Wang, P., et al., Synthesis of phospholipid-inhibitor conjugates by enzymatic transphosphatidylation with phospholipase D. J. Am. Chem. Soc., 1993. 115: p. 10487-10491. |
|
17. |
Hostetler, K.Y., et al., Synthesis and antiretroviral activity of phospholipid analogs of azidothymidine and other antiviral nucleosides. J. Biol. Chem., 1990. 265(11): p. 6112-7. |
|
18. |
van Wijk, G.M., K.Y. Hostetler, and H. van den Bosch, Lipid conjugates of antiretroviral agents: release of antiretroviral nucleoside monophosphates by a nucleoside diphosphate diglyceride hydrolase activity from rat liver mitochondria. Biochim. Biophys. Acta, 1991. 1084(3): p. 307-10. |
|
19. |
van Wijk, G.M., et al., Cytidine diphosphate diglyceride analogs of antiretroviral dideoxynucleosides: evidence for release of dideoxynucleoside-monophosphates by phospholipid biosynthetic enzymes in rat liver subcellular fractions. Biochim. Biophys. Acta, 1991. 1086(1): p. 99-105. |
|
20. |
van Wijk, G.M., K.Y. Hostetler, and H. van den Bosch, Antiviral nucleoside diphosphate diglycerides: improved synthesis and facilitated purification. J. Lipid Res., 1992. 33(8): p. 1211-9. |
|
21. |
van Wijk, G.M., et al., Synthesis, characterization and some properties of dideoxynucleoside analogs of cytidine diphosphate diacylglycerol. Biochim. Biophys. Acta, 1992. 1165(1): p. 45-52. |
|
22. |
Hostetler, K.Y., et al., Greatly enhanced inhibition of human immunodeficiency virus type 1 replication in CEM and HT4-6C cells by 3'-deoxythymidine diphosphate dimyristoylglycerol, a lipid prodrug of 3'-deoxythymidine. Antimicrob. Agents Chemother., 1992. 36(9): p. 2025-9. |
|
23. |
van Wijk, G.M., et al., Spontaneous and protein-mediated intermembrane transfer of the antiretroviral liponucleotide 3'-deoxythymidine diphosphate diglyceride. Biochemistry, 1992. 31(25): p. 5912-7. |
|
24. |
Hostetler, K.Y., et al., Acyclovir diphosphate dimyristoylglycerol: a phospholipid prodrug with activity against acyclovir-resistant herpes simplex virus. Proc. Natl. Acad. Sci. U S A, 1993. 90(24): p. 11835-9. |
|
25. |
van Wijk, G.M., et al., Synthesis and antiviral activity of 3'-azido-3'-deoxythymidine triphosphate distearoylglycerol: a novel phospholipid conjugate of the anti-HIV agent AZT. Chem. Phys. Lipids, 1994. 70(2): p. 213-22. |
|
26. |
Kato, T., et al., Conformational studies of amphipathic a-helical peptides containing an amino acid with a long alkyl chain and their anchoring to lipid bilayer liposomes. Biochim. Biophys. Acta, 1991. 1063: p. 191-196. |
|
27. |
Yager, P. and P.E. Schoen, Formation of tubules by a polymerizable surfactant. Mol. Cryst. Liq. Cryst., 1984. 106: p. 371-381. |
|
28. |
Schoen, P.E. and P. Yager, Spectroscopic studies of polymerized surfactants: 1,2-bis(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine. J. Polym. Sci.;Polym. Phys. Ed., 1985. 23: p. 2203-2216. |
|
29. |
Yager, P., et al., Structure of lipid tubules formed from a polymerizable lecithin. Biophys. J., 1985. 48: p. 899-906. |
|
30. |
Yager, P., et al., Two mechanisms for forming novel tubular microstructures from polymerizable lipids. Biophys. J., 1986. 49(2): p. 320. |
|
31. |
Chappell, J.S. and P. Yager, Electrolyte effects on bilayer tubule formation by a diacetylenic phospholipid. Biophysical Journal, 1991. 60(4): p. 1-14. |
|
32. |
Georger, J., et al., Helical and tubular microstructures formed by polymerizable phosphatidylcholines. J. Am. Chem. Soc., 1987. 109: p. 6169-6175. |
|
33. |
Markowitz, M. and A. Singh, Self-assembling properties of 1,2-diacyl-sn-glycero-3-phosphoydroxyethanol: a headgroup-modified diacetylenic phospholipid. Langmuir, 1991. 7: p. 16-18. |
|
34. |
Fuhrhop, J.H., et al., Long-lived micellar n-alkylaldonamide fiber gels. solid-state NMR and electron microscopic studies. J. Am. Chem. Soc., 1990. 112: p. 4307-12. |
|
35. |
Fuhrhop, J.H., et al., Lipid bilayer fibers from diastereomeric and enantiomeric n-octylaldonamides. J. Am. Chem. Soc., 1988. 110: p. 2861-7. |
|
36. |
Nakashima, N., et al., Helical superstructures are formed from chiral ammonium bilayers. Chem. Lett., 1984: p. 1709-1712. |
|
37. |
Nakashima, N., S. Asakuma, and T. Kunitake, Optical microscopic study of helical superstructures of chiral bilayer membranes. J. Amer. Chem. Soc., 1985. 107: p. 510-512. |
|
38. |
Yamada, K., et al., Formation of helical super structure from single-walled bilayers by amphiphiles with oligo-L-glutamic acid-head group. Chem. Lett., 1984. 10: p. 1713-1716. |
|
39. |
Ihara, H., et al., Amphiphiles with polypeptide-head groups. 7. Relationship between formation of helical bilayer membranes and chemical structures of dialkyl amphiphiles with polypeptide-head groups. Kobunshi Ronbunshu, 1991. 48(5): p. 327-34. |
|
40. |
Shimizu, T. and M. Hato, Self-assembling properties of synthetic peptidic lipids. Biochimica et Biophysica Acta, 1993. 1147: p. 50-58. |
|
41. |
Ihara, H., et al., Production of helical bilayer membranes from L-glutamic acid derivatives with bis(dodecylamide) groups and their specific optical activity. Nippon Kagaku Kaishi, 1990. 10: p. 1047-53. |
|
42. |
Nakashima, N., et al., Dynamic transformation of the morphology of dialkylammonium bilayer aggregates. Chem. Lett., 1984: p. 227-230. |
|
43. |
Archibald, D.D., Structural studies of high aspect-ratio self-assembled lipid microstructures with the use of microscopy and FT-NIR-Raman spectroscopy, . 1990, University of Washington. |
|
44. |
Archibald, D.D. and P. Yager, Microstructural polymorphism in bovine brain galactocerebrosides and its two major subfractions. Biochemistry, 1992. 31(37): p. 9045-9055. |
|
45. |
Johnston, D. and D. Chapman, The properties of brain galactocerebroside monolayers. Biochim. et Biophys. Acta, 1988. 937: p. 10-22. |
|
46. |
Naito, M., K. Takahashi, and H. Hojo, An ultrastructural and experimental study on the development of tubular structures in the lysosomes of Gaucher's cells. Lab. Invest., 1988. 58(5): p. 509-598. |
|
47. |
Fisher, E.R. and H. Reidbord, Gaucher's disease: pathogenetic considerations based on electron microscopic and histochemical observations. American Journal of Pathology, 1962. 41(6): p. 679-686. |
|
48. |
Yunis, E.J. and R.E. Lee, Tubules of globoid leukodystrophy: a right-handed helix. Science, 1970. 169: p. 64-66. |
|
49. |
Archibald, D.D. and S. Mann, Template mineralization of self-assembled anisotropic lipid microstructures. Nature, 1993. 364(29 July): p. 430-433. |
|
50. |
Archibald, D.D. and S. Mann, Self-assembled microstructures from 1,2-ethanediol suspensions of pure and binary mixtures of neutral and acidic biological galactosylceramides. Chem. Phys. Lipids., 1994. 69(1): p. 51-64. |
|
51. |
Archibald, D.D. and S. Mann, Structural studies of lipid fibers formed by sphingosine. Biochim. Biophys. Acta, 1993. 1166(2-3): p. 154-62. |
|
52. |
Servuss, R.M., Helical Ribbons of Lecithin. Chem. Phys. Lip., 1988. 46: p. 37-41. |
|
53. |
Konikoff, F.M., et al., Filamentous, helical, and tubular microstructures during cholesterol crystallization from bile. J. Clin. Invest., 1992. 90(Sept.): p. 1155-1160. |
|
54. |
Chung, D.S., et al., Elastic free energy of anisotropic helical ribbons as metastable intermediates in the crystallization of cholesterol. Proc. Natl. Acad. Sci. USA, in press. |
|
55. |
Zhong-can, O.-Y. and L. Jixing, Theory of helical structures of tilted chiral lipid bilayers. Physical Review A, 1991. 43(12): p. 6826-36. |
|
56. |
Zhong-can, O.-Y. and L. Ji-Xing, Helical structures of tilted chiral lipid bilayers viewed as cholesteric liquid crystals. Physical Review Letters, 1990. 65(13): p. 1679-82. |
|
57. |
Chappell, J.S. and P. Yager, A model for crystalline order within helical and tubular structures of chiral bilayers. Chemistry and Physics of Lipids, 1991. 58: p. 253-258. |
|
58. |
Chappell, J.S. and P. Yager, Electrostatic interactions within helical structures of chiral lipid bilayers. Chemical Physics, 1991. 150: p. 73-79. |
|
59. |
Helfrich, W., Helical bilayer structures due to spontaneous torsion of the edges. J. Chem. Phys., 1986. 85(2): p. 1085-1087. |
|
60. |
de Gennes, P.-G., Physique des Surfaces et des Interfaces. C. R. Acad. Sc. Paris, 1987. 304(II7): p. 259-263. |
|
61. |
Helfrich, W. and J. Prost, Intrinsic bending force in anisotropic membranes made of chiral molecules. Physical Review A, 1988. 38(6): p. 3065-3068. |
|
62. |
Selinger, J.V. and J.M. Schnur, Theory of chiral lipid tubules. Physical Review Letters, 1993. 71(24): p. 4091-4094. |
|
63. |
Schnur, J.M., et al., Metal clad microstructures, , D.o.t.N.G. Inc., Editor. 1990, USA: 4911981. |
|
64. |
Behroozi, F., et al., Interaction of metallized tubules with electromagnetic radiation. J. Appl. Phys., 1990. 68(7): p. 3688-3693. |
|
65. |
Chappell, J.S. and P. Yager, Formation of mineral microstructures with a high aspect ratio from phospholipid bilayer tubules. J. Mat. Sci. Lett., 1992. 11: p. 633-636. |
|
66. |
Schnur, J.M., R. Price, and A.S. Rudolph, Biologically engineering microstructures: controlled relase applications. Journal of Controlled Release, 1994. 28: p. 3-13. |
|
67. |
Mann, S., et al., Crystallization at inorganic-organic interfaces: Biominerals and biomimetic synthesis. Science, 1993. 261(3 Sept.): p. 1286-1292. |
|
68. |
Lipkin, R., Crystal coated lipids promise new materials. Science News, 1993. 155(5)(July 31): p. 69. |
|
69. |
Degani, R., Lipids and minerals form novel composite microstructures. C&EN, 1993(August 9): p. 19-20. |
|
70. |
Rudolph, A.S., et al., Technological development of lipid based tubule microstructures, in Biotechnological Applications of Lipid Microstructures, B.P. Gaber, J.M. Schnur, and D. Chapman, Editors. 1988, Plenum Publishing Corp.: New York. p. 305-320. |
|
71. |
Cliff, R.O., B.J. Spargo, and A.S. Rudolph. The use of lipid microcylinders as release vehicles: release rates of growth factors and cytokines. in Fourth World Biomaterials Conference. 1992. Berlin, FRG. |
|
72. |
Rudolph, A.S., et al., Biocompatibility of lipid microcylinders: effect on cell growth and antigen presentation in culture. Biomaterials, 1992. 13(15): p. 1085-1092. |
|
73. |
Masure, S. and G. Opdenakker, Cytokine-mediated proteolysis in tissue remodeling. Experientia, 1989. 45: p. 542-549. |
|
74. |
Sandset, P.M. and U. Abildgaard, Extrinsic pathway inhibitor--the key to feedback control of blood coagulation initiated by tissue thromboplastin. Haemostasis, 1991. 21(4): p. 219-39. |
|
75. |
Scharpe, S., et al., Proteases and their inhibitors: today and tomorrow. Biochimie, 1991. 73(1): p. 121-6. |
|
76. |
Uchio, H., et al., Studies on the special type surfactants. XIII. preparation and properties of oligopeptides. Yukagaku, 1984. 33(8): p. 510-517. |
|
77. |
Plant, A.L., D.M. Benson, and G.L. Trusty, Probing the structure of diacetylenic phospholipid tubules with fluorescent lipophiles. Biophys. J., 1990. 57: p. 925-933. |
|
78. |
Jain, M.K., et al., Kinetics of interfacial catalysis by phospholipase A2 in intravesicle scooting mode, and heterofusion of anionic and zwitterionic vesicles. Biochim. Biophys. Acta, 1986. 860: p. 435-447. |
|
|
Return to <Yager's Home Page or to Tubule Project Page> |
|