|
Bioengineering Department, Box 355061, University of Washington, Seattle, WA 98195, USA |
 |
Silk Protein Project
|
|
Bioengineering Department, Box 355061, University of Washington, Seattle, WA 98195, USA |
|
This project was funded for over three years by the National Science Foundation, originally with Christopher Viney of UW as Principal Investigator, and Yager as Co-PI. Viney has moved to Oxford (UK) and funding has recently ended, and a single graduate student is finishing the project as her Ph.D. project.
Paul Yager, PI
Kimberly Carlson (formerly Trabbic), Ph.D. Candidate
Although both industry and nature are able to produce high-performance polymers, nature has evolved far superior methods for processing polymers into materials. These methods allow nearly optimal utilization of the mechanical properties of the polymer chains, but employ simpler processing conditions that are chemically and physically far milder than those used for man-made high strength polymers. If we can learn how the natural processing results in enhanced physical properties, we should be able to use that knowledge in the development of superior materials, whether the polymers of which they consist are of man-made or of natural origin. Materials of biological origin have the additional significant advantages of intrinsic compatibility with biological systems and bio degradability.
An especially clear-cut example of the contrast between biological and synthetic approaches is the spinning of silk. The processability of silk from solution to fiber is intimately related to the molecular nature of the polymer. While synthetic polymers are made of one or two repeating monomer units polymerized to a broad range of lengths, biological polymers such as silk fibroin are identical molecules of great complexity made up of nearly 20 different amino acid monomers. A wide range of tailorable mechanical properties can be achieved by varying the sequence of amino acids. Frame silk and capture threads in a spider web differ only in amino acid composition.
Spiders and insects have been able to spin silks for at least 380 million years [1], so there has been time for a high degree of process optimization. The breaking strength and elastic modulus of spider dragline silk (up to 2 GN.m-2 and 30 GN.m-2, respectively [2-4]) exceed the values for highly drawn nylon (0.7 GN.m-2 and 2.4 GN.m-2, respectively [5]). Thus, an industrial product that is spun at elevated temperatures, and that requires a subsequent draw ratio of several hundred per cent for optimum tensile properties, is matched by a natural material spun into fibers under ambient conditions without the need for a post-spinning draw. If we compare spider dragline silk to Kevlar, we find that the silk has lower strength and stiffness, but ten times greater toughness [3]. While Kevlar is spun from solution in hot fuming sulfuric acid, natural silk fibers are produced at room temperature and from aqueous solutions.
The recent demonstration of cloning and expression of silk fibroins including spider dragline silk [4,6-7] represents an exciting scientific opportunity and a technological challenge. We can now obtain biosynthetic spider dragline fibroin in research quantities from Dr. D.L. Kaplan of the U.S. Army Research, Development & Engineering Center (see attached letter). This will allow us to spin fibers from precisely defined solutions through orifices of our own design. The technological challenge is that if one is to make commercial use of the biosynthetic dragline silk, one must develop processing methods for the fibroin solutions that equal or improve upon spinning by spiders. Silks and similar polymers may also be formed into structures other than fibers &endash; for example bubbles can be blown with silkworm silk [8]. Indeed, in principle any structure can be made that allows ready removal (evaporation) of solvent (usually water) during its formation. The three-dimensional b-sheet structure of silks may also be the basis for better structural properties in fibers and films derived from silks, compared to fibers and films formed by high strength synthetic liquid crystalline polymers that typically exhibit poor resistance to crack propagation in the direction of molecular alignment. "Spinning", as opposed to extrusion, of polymeric sheets should be possible with silks.
While we have a fairly good understanding of how simple synthetic polymer chains align in a drawn fiber, we have only the most rudimentary understanding of how the highly complex fibroin molecules self-organize to form a high strength polymer, and how they manage to do so under the mildest of conditions. The materials science issues cannot be separated from the questions of the three-dimensional structure of the fibroin molecule. The problem does not fit within the boundaries of a single discipline &endash; it brings together materials science, the biophysics of protein folding, molecular biology and the biology of organisms. We propose to address this problem by combining background skills in materials science (Viney) and physical chemistry (Yager) in the interdisciplinary research environment provided in the Molecular Bioengineering Program of the Center for Bioengineering at the University of Washington.
We will study natural products of both silkworms and spiders and genetically engineered (cloned and expressed) synthetic fibroin. If we can understand how the spinning process works, we can use this knowledge to improve processing in existing man-made polymers, allow utilization of fermentation-derived silks, and extend the use of silks and silk-like polymers to structures other than fibers.
Much work has been done in the last decade to attempt to understand the relationship between the molecular structure of silks and their mechanical and physical properties. An excellent review will shortly be available [4]. The fibroin that is the only polypeptide in the dragline silk of Nephila clavipes spiders has a molecular weight of about 320 kD, which, if fully extended could be nearly 0.5 µm long. The recent partial sequencing of this silk [9] has greatly added to our understanding of the structure of these molecules, and its cloning and expression [4,6-7] affords us the potential of obtaining large quantities of pure spider dragline fibroin for in vitro experimentation. Xu et al. find the sequence to be quite different from Bombyx mori (silkworm) silk. Most strikingly, the molecules in both types of silk consist of an alternating sequence of segments likely to form b-sheets and segments likely to remain in random coil or form a-helix structures; yet, the lengths of these segments are much greater in silkworm silk. Spider silk fibroin is much less regular in sequence than silkworm silk, which may explain why spun spider silk contains a greater proportion of amorphous material.
A good working model for the microstructure of spider dragline silk is provided by Gosline et al. [3]. Their view of the silk fiber is as a micro composite of small anisotropic crystallites of b-sheet embedded in a matrix of amorphous rubbery polypeptide. The crystalline domains are presumed to have an axial ratio of about 5 and occupy only about 30% of the volume of the dry silk. The rest is seen as occupied by polypeptide segments of about 15 amino acids that are glassy when dry and rubbery when swollen in distilled water. The b-sheet segments seem impervious to swelling in water [10-11]. The long fibroin molecules must extend through many crystallites and interconnect them through the amorphous regions. The 34-amino acid repeat found by Xu and Lewis in the sequence of spider dragline [9] includes regions high in alanine that may correspond to the amorphous regions suggested by Gosline.
Before spinning, the fibroin molecule as synthesized by either silkworm or spider is soluble, forming a non-Newtonian fluid at concentrations exceeding 25% by weight [12-13]. There is reputed to be a small amount of a-helix in the structure, but it is otherwise without detectable regularity of secondary structure. Its solutions are moderately viscous, but are easily handled and stored at room temperature. When a critical shear stress is applied to concen trated fibroin solutions, the molecules undergo a conformational change similar to partial denaturation, generating the dense form found in silk fiber. While spider dragline contains domains of b-sheet and random coil [3], silkworm fiber is almost all b-sheet [13]. The b-sheet regions are rigid and inextensible. When the completed dragline is stretched, some of the remaining random coil converts transiently into a-helix in a process that may be responsible for the dragline's superior elasticity over silkworm silk [11]. The denaturation has the characteristics of a phase transition, but it is not understood why or how it happens.
We have a general knowledge of the overall secondary structural compositions and orientations of the pre- and post-spinning materials. However, nothing is known of the change in secondary structure that occurs during spinning. The changes that occur are as drastic as those in complete denaturation of a globular protein, and similar theoretical techniques could be applied to understand this problem. The only difference is that this process only occurs in the presence of shear and at high enough concentration that intermolecular interactions probably dominate over intramolecular ones. The relationship between concentration and the critical shear rate needed to trigger the conversion to b-sheet in natural silk is not known. A critical uniaxial extension rate of approximately 1 cm/second is quoted [14-15] for silkworm glands that have been washed to remove non-fibroin contents. This is approximately the rate at which silkworms spin fiber in nature; however, such comparison is flawed until it can be made on the basis of similar shear rates.
High strength and stiffness indicate a microstructure in which there is significant molecular extension and alignment. In conventional polymer fiber processing, both the extension and alignment introduced at the spinnerets are lost during the time taken for the fiber to solidify. A post-spinning draw is required to recover these properties. Liquid crystalline polymers have become increasingly attractive to industry [16] because the molecules are tailored to have an extended conformation in the processing environment, and they align spontaneously into domains that typically are several microns across. Thus it is only necessary to align whole domains, and not individual molecules, to generate global alignment in a fiber. Still, elevated temperatures and/or hazardous solvents are required to enable processing.
Using transmitted polarized light microscopy, we have determined that B. mori secretions, and the major ampullate (dragline and frame silk), tubuliform (cocoon silk) and flagelliform (capture thread) secretions of N. clavipes spiders, form nematic liquid crystalline phases when water is lost by evaporation [17-18]. Thus we now have a qualitative understanding of
Indeed, ease of processing appears to be the principal benefit of liquid crystalline order, since the different types of silk exhibit a wide range of strength and stiffness.
We observed that the nematic domain size varies significantly among the silks studied. For example, the microstructural scale of spider dragline secretion is an order of magnitude finer than that of the silkworm secretion [21]. This suggests that the "rods" responsible for nematic behavior in the Bombyx silk secretion should be significantly longer than those in the Nephila dragline secretions. But a simple computer prediction of residues with the greatest potential for a-helix formation finds that the average a-helix length is slightly greater in representative sequences of Nephila dragline silk, suggesting that a-helices (i.e. rods at the scale of molecular segments) are not the rod-like structural unit responsible for liquid crystalline behavior in these materials [18]. Because the liquid crystalline phase of silk secretions appears even in the absence of shear, and the molecular conformation in the absence of shear does not appear to depend on concen tration [22], we suspect that the "rods" must result from the aggregation of protein molecules as their concentration increases.
|
|
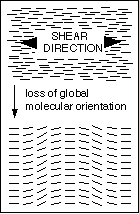
Figure 1 Top: global molecular orientation induced in drawn or sheared nematic material. Bottom: the spatially periodic misalignment of molecular orientation that typically develops in the fluid nematic after shear has ceased. The minimum microstructural periodicity is limited by the elastic constants of the nematic phase, and can therefore be resolved optically. |
Our immediate objective is to determine the nature of the changes in molecular conformation and the associated evolution of microstructure that accompany silk fiber spinning in nature and in biosynthetic silk. The longer term goal is to provide the scientific basis for improved processing of fibroins and other polymers into fibers, sheets, and other morphologies.
We will be concerned with how the size, shape, and orientation of rigid and compliant domains within the fiber, and also the resultant mechanical properties, are determined by the processing conditions. Studies will be conducted on silk spun in vivo, and on both natural and genetically engineered silk processed in vitro. The effect of the following processing variables will be studied: draw rate, shear rate (in vitro only), spinneret geometry, temperature, humidity, salt concen tration, presence of other molecules (such as surfactants). While such a study will necessarily increase our basic understanding of protein folding and the natural silk fiber spinning process, we believe that it will be of great practical value if the knowledge is applied to develop schemes for processing high performance polymeric materials in benign environments. Also, the proposed work should add to the growing knowledge base for use in designing new polypeptide-based materials.
We will employ several physical techniques for the elucidation of the molecular events in fibroin spinning. These include optical microscopy, Raman spectroscopy, and X-ray diffraction. We intend to employ the set above insofar as is possible to monitor the spinning process itself, and thereby be able to bridge the gap between our knowledge of the molecular biology of spider silk and our understanding of the drawing of liquid crystal polymers. We will use a polymer modelling package (CERIUS, from Polygen - Molecular Simulations) for interpreting x-ray and electron diffraction data. The complete sequence of one spider dragline fibroin, along with biosynthetic spider fibroin itself, will be provided by Dr. David Kaplan of the Natick RD&E Center (see attached letter). As experimental data from microscopy, spec troscopy and diffraction are obtained, we will attempt to use interactive computer graphics model building techniques and the secondary structure information to construct plausible three-dimensional molecular structures that are consistent with the observed unit cell packing data.
Three of the items of equipment that we will use have been custom-built and/or uniquely configured: a Raman spectrometer, a shearing stage for use with a polarizing light microscope, and an apparatus for drawing silk either from live spiders and silkworms or from solutions of fibroin. These items, and their role in the proposed studies, are described in more detail below.
Among the best ways to study protein secondary structure in solution is Raman spec tros copy. The technique has been underutilized primarily because of the expense and resultant scarcity of high quality equipment. Changes in f and u angles, in hydrogen bonding, and electrostatics of the environment all affect the vibrations of the peptide bonds, giving rise to a sensitivity of the amide normal modes to the secondary structure of the protein [30]. In addition, subtle changes in sidechain vibrations can also be correlated with alterations in conformation or solvent. Both Fourier transform infrared spectroscopy (FTIR) and Raman scattering (both conventional and NIR-FTIR Raman) have strengths and weaknesses in studying polypeptide structure [31], but technical advances have recently greatly enhanced the utility of Raman spectroscopy. Nonspecific luminescence, for decades the nemesis of the technique, can be greatly reduced by using a red or near infrared laser sources. The new two-dimensional CCD detectors have excellent response in the far red, dynamic range of up to 105, and, when cooled to lN2 temperatures, better quantum efficiency than cooled PMTs with almost no noise (1 count per pixel per hour!). Because each of the thousands of CCD pixels has such high sensitivity, spectra that may have taken 12 hours to collect a decade ago may now be collected in 30 seconds, and smaller samples can be used at lower laser powers. For visible and some NIR frequencies, dispersive CCD-based systems are superior to any other detector type, including interferometers [32].
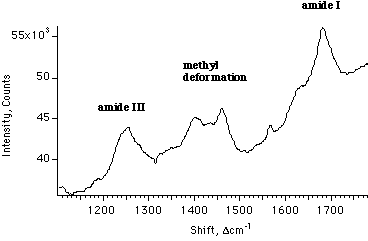
Figure 2. A Raman spectrum obtained from a single 2 µm thick strand of spider dragline silk. The laser beam was vertical (parallel to the slits) and focussed to a beam waist of approximately 50 µm; the fiber was held at a 15° angle tilted toward the monochromator slit. Collection time 20 sec, 300 mW 5145Å laser power, 100 µm slit.
Recent successful competition by the Center for Bioengineering for a grant for enhancement to education in Bioengineering from the Whitaker Foundation has allowed us to purchase a unique state-of-the-art CCD-based Raman spectrometer (cost ~$130,000). It has just become fully operational, although improvements to the optics and software for data handling are ongoing. The instrument is based on a 0.5 meter single monochromator (SPEX 500M) with moderate resolution but high throughput, coupled to a 385 by 578 pixel Astromed MPP CCD detector operated at 136°K. The detector's low background of 1 photoelectron per pixel per hour allows rapid collection of extremely low signals; spectral resolution is quite adequate for all biologically relevant samples in solution. The instrument is controlled by SPEX software running on an IBM 386 clone computer. The system is operated in a manner in which the slit image is projected along the short axis of the detector (vertical), and the grating disperses the spectrum horizontally. As the CCD area can currently be electronically sampled as if it were 1 to 4 independent hori zontal detectors, it is possible to simultaneously collect four independent spectra from segments at different heights along the slit axis. Because there is only one slit in the instrument, some form of filter to remove Rayleigh-scattered light is required for scattering samples such as fibers. We are currently using an interference filter, but have just pur chased a holographic filter, which offers the possibility of flatter response and retention of the important reference bands provided by water Raman scattering at ~3300 cm-1. The current light source is a 4 watt Lexel tunable argon ion laser, and the equipment is mounted on a Newport 4'x6' floating optical table. An example of the remarkable sensitivity of the system is seen in Figure 2. To use a source redder than 5145Å to avoid luminescence problems we have purchased a 10 mW HeNe and holographic filter combination, and will be assessing the adequacy of this source.
The most important recent development in protein vibrational spectroscopy has been the application of mathematical methods such as factor analysis and partial least-squares to quantification of secondary structure. The methods are similar to ones applied to analysis of circular dichroism spectra, but rely instead on the amide I region, and often the amide II and III regions as well. While monitoring of the peak frequency of the amide I region continues to be adequate for simple differentiation between predominantly a-helical structure and predominantly b-sheet or random structure [31,33], the bands strongly overlap, so more complex mixtures of secondary structure produce complex amide I bands that require more sophisticated analysis. Methods have been developed for the interpretation of infrared [34-36] and Raman [37-39] amide I regions, and comparable accuracies are obtained under ideal conditions with both spectroscopic techniques. The best solution to date for the problem using Raman spectroscopy has been made by Williams [37-38], whose programs are now available for use by our laboratory. His technique relies on assuming that the amide I region of the spectrum of the protein in question can be treated as a linear combination of spectra from their component secondary structures.
Williams' software provides a method for taking Raman data from a 300 cm-1 wide region of the spectrum, subtracting luminescence background and sidechain peaks, and then quantifying the percentages of a-helix (ends vs ordered cores), b-sheet, b-turn, and disordered structure. We have used Williams' method to predict the secondary structure of a protein nearly as large as fibroin, the 260 kD nicotinic acetylcholine receptor from Torpedo californica, and the predictions have held up well [40]. Raman has been used for the determination of silkworm fibroin secondary structure both before and after spinning [15], and some excellent spectra have been recorded [14]. The spectra of pre- and post-spinning fibroin are strikingly different and easily distin guished, but as yet no one has applied a quantitative analysis to them.
As the laser is polarized, comparison of Raman scattering parallel and perpendicular to the laser polarization can be used to determine the degree and direction of orientation of the groups (such as the amide bonds) responsible for observed scattering. Not only can the orientation of the peptide backbone be determined, but also that of specific side chains with distinctive vibrational bands, such as the aromatic amino acids. In addition, sample size is not generally a problem. Water and other solvents have Raman-active vibrations, so the technique can also be used to determine the concentration of solvents in a sample. As the Raman effect is nearly instantaneous (~10-14 sec), as long as an adequate signal-to-noise ratio (SNR) can be obtained, Raman allows measurement of the kinetics of protein folding. It could also be an excellent tool for monitoring production of fibers in real time as these are drawn. As Raman spectrometers are essentially very sensitive fluorimeters, they can also be used with fluorescent probes to monitor sample orientation, polarity, viscosity, and pH.
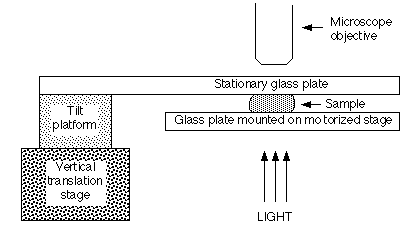
We have designed a parallel plate shearing apparatus that can be fitted to a Leitz Laborlux 12 POL transmitted polarized light microscope (see Figure 3). The device has been constructed and is in use [29,41]. It provides controlled shear of a sample between two glass plates: a stationary upper plate and a motorized lower plate. The upper plate is cantilevered from a Newport Model 37 tilt platform mounted on top of a Newport Model 415 vertical translation stage. Two micrometer tilt adjustments on the platform allow accurate positioning of the plane of the upper plate. The translation stage controls the vertical displacement of the upper plate, with an accuracy of 0.5 µm. The lower plate is attached to a motorized Leitz Model 1089 x-y stage, which travels a maximum of 7.6 cm in the x direction and 4.6 cm in the y direction. Joystick control provides plate velocities of up to 9 cm/sec.
|
|
Figure 3
Schematic representation of stage used to shear thin samples under a transmitted light microscope.
|
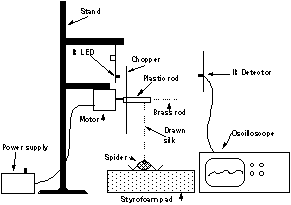
The existing apparatus for forced silking consists of seven sub-components: stand, electric motor, IR LED assembly, IR detector, power supply for the motor, stereomicroscope, and spider restraint (see Figure 4). This equipment is based on an earlier design in the literature [42].
The electric motor is a low speed (0-200rpm) geared motor and is used to withdraw silk from the spider. A 1/4 inch plexiglas rod with a hole drilled along its axis is pressure fit to the shaft. A 1/16 inch hole is drilled in the other end of the rod, to allow insertion of a 1/16 inch diameter rod for use at low draw rates. Also attached to the 1/4 inch rod is a "chopper". This is a circular piece of cardboard with 12 evenly-spaced holes near the perimeter, that periodically interrupts the IR LED beam and is used for RPM calibration. The IR photodetector is connected to an oscilloscope. A variable-voltage power supply is constructed from a 12V AC, 500mA transformer, connected to a full-wave diode rectifier. The output is controlled and smoothed by a capacitor and resistor attached in parallel with a potentiostat. Spiders are restrained upside down by two rubber bands that are pinned to a closed-cell styrofoam block; each band runs along one side of the spider and is used to trap four legs near the point where they join the cephalothorax.
|
|
Figure 4
Schematic representation of the apparatus used for silking.
|
The formation of liquid crystalline phases in natural silk secretions will be monitored by transmitted polarized light microscopy of thin specimens held between glass slides
By noting what microstructures are formed, and how the observed contrast changes when the crossed polars are rotated, we will identify any liquid crystalline phases formed. This work will be performed on frame silk, capture thread, and cocoon silk for the three species of spider about which the most is already known: N. clavipes, A. diadematus and A. sericatus. The study will not include attachment pad silk or prey-wrapping silk, because the relevant glands are too small to conveniently dissect from the spiders. Because we cannot be certain that the fibroin composition in two different specimens of the same species will be identical, we will need to compare results obtained from several specimens.
By using large droplets supported between glass microscope slides, we anticipate that water loss by evaporation should lead to a situation where the perimeter is a crystalline solid and the center is an isotropic fluid. Then the critical concentrations required for a stable liquid crystalline phase could be determined by measuring the relative intensities of protein and water in Raman spectra from different positions along a radius of the droplet. If it is not possible to obtain a large enough droplet (i.e. if the perimeter is still liquid crystalline when the center of the droplet has also become liquid crystalline), we would perform repeated measurements of the concentration profile, and allow intervening time for the sample to lose water. We will perform control experiments with regenerated solutions of silkworm silk, which is readily available in sufficient quantities to enable the preparation of solutions with known concentration.
From the critical concentration of polymer required to form a single anisotropic phase, we will estimate the axial ratio of the rodlike structures that are the basis of the nematic order in this system. We will assume the validity of Flory's models [43] relating critical concentration to axial ratio for athermal systems. Athermal behavior is assumed on the basis of our observations that no biphasic (mixture of isotropic and nematic) material is resolved between the isotropic interior and liquid crystalline perimeter of samples investigated in our preliminary studies. From this, we deduce that biphasic behavior is restricted to a narrow concentration range, and therefore that the material has the thermodynamic characteristics of an athermal system.
Samples with a known concentration profile will be heated or cooled under the microscope on a calibrated heating/freezing stage (LinkAm THS 600). By noting how the interface between crystalline and nematic material &emdash; or between isotropic and nematic material &emdash; is displaced, we will determine how the critical concentrations for stabilizing a liquid crystalline phase depend on temperature. In this way, we will build up the phase diagrams for a variety of natural silk secretions in the vicinity of room temperature. The lower temperature limit for these experiments is set by the freezing point of the solutions. The upper limit is determined by observing whether the concentration profile in the sample changes significantly during the experiment; we will subsequently check for such changes by re-measuring the concentration profile at ambient temperature.
Our experiments will be repeated on solutions of genetically engineered spider dragline fibroin, from which we will attempt to determine what requirements of pH and/or ionic strength must be met to make this material behave like the natural silk secretion. This will enable us to develop a composition that can be used for in vitro studies of the effect of different processing variables on the formation of silk fiber.
The light microscopy studies described in this task will therefore enable us to answer the following questions:
Samples will be observed under the microscope on the shearing stage described above. The parallel plate geometry of the shearing stage will provide a uniform shear rate through the sample thickness, which represents a convenient simplification relative to fiber pulling through a spinneret. Also, obser vations of samples between crossed polars will allow us to determine whether the periodic microstructure in Figure 1 above develops upon cessation of shear. In this way we hope to identify combinations of concentration and shear rate that either prevent this processing defect from forming, or that at least retard it over a timescale that is significantly longer than the in vivo solidification time.
Initially, our experiments with the shearing stage will look at both dragline secretion from N. clavipes and solutions of its genetically engineered counterpart. If we can obtain reasonable agreement between the behavior of both materials under a common set of processing conditions, we will use the genetically engineered silk as a model for studying at the effect of varying the processing conditions. If the agreement is poor, we will work with solutions of natural silk secretion only.
The experiments with the shearing stage will provide answers to the following questions:
We will use electron microdiffraction [44] to distinguish between crystalline and amorphous regions. The scale of the purported microstructure [3] is easily within the resolution capabilities of this technique. We will examine sequential slices microtomed from naturally spun fibers, natural fibers produced by forced silking, fibers drawn in vitro from natural silk secretion, and fibers drawn from genetically engineered silk secretion.
The TEM experiments are expected to answer the following questions:
Forced silking at different rates will be accomplished with the equipment described above. Molecular orientation will be measured from the azimuthal spread of inter molecular diffraction maxima obtained on a flat film with a Warhus x-ray camera [45] (X-rays will be used instead of TEM in this experiment because we want to conveniently sample the average molecular orientation in the bulk fiber). The optical birefringence will also be measured by using a transmitted polarized light microscope equipped with a compensator. From a plot of birefringence vs molecular orientation, we will determine the intrinsic birefringence by extrapolation. Then we will be able to determine the degree of orientation in naturally spun silk by simply measuring its birefringence. The average molecular orientation in silk fiber may depend on the ambient humidity (which we will measure), and it will depend on the average shear rate through the fiber cross-section. This in turn depends on both the extension rate and the diameter of the orifice through which the silk is drawn. We will draw natural silk secretion through artificial spinnerets at the extension rates typically found in nature. By correlating birefringence measurements (average degree of molecular orientation) with spinneret size, we will obtain a measure of the true shear rates that are used in nature.
The results of these experiments will answer the following questions:
It is known that the composition of the fibroin solution and the orientation and secondary structure of the fibroin molecules themselves change during the process of spinning liquid silk into a fiber. We will study these changes by performing Raman (and possibly fluorescence) spectroscopy on natural and artificial single silk fibers during spinning. Samples will include silk fibers spun from spiders and silkworms, as well as from extracted fibroin and from genetically engineered fibroin using artificial spinnerets. This will allow the first temporal and spatial profiling of the water content, structure and molecular orientation along silk fibers during conversion from the random coil fibroin solution.
The spectroscopic studies are designed to provide answers to the following questions:
To quantify the fibroin secondary structure, we will use the aforementioned method developed by Prof. R.W. Williams at USUHS in Bethesda [38]. The package from Williams also includes his basis set of Raman spectra of 15 proteins whose secondary structure is known from crystallographic studies. We are now in the process of converting his FORTRAN code to a form that will work on one of our local VAX machines. Each Raman system is optically unique, so to use Williams' method we must first normalize his basis set to our instrument by running at least one reference spectrum such as lysozyme. The normalization for one protein can be applied to the rest of the imported basis set spectra.
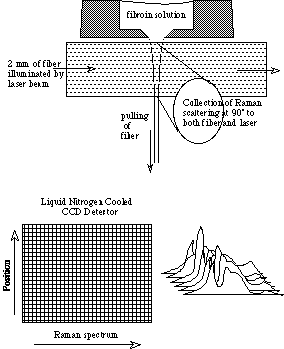
With the current Nikon f/1.2 lens used for collection of scattered light, each detector pixel covers a vertical height at the sample of approximately 7 µm, which corresponds to the spatial resolution of the system (see Figure 5). As this is roughly the diameter of the silk fibers that we will be observing, this is adequate spatial resolution for our purposes. However, while it is possible to collect spectra from a single row of pixels, the S/N ration will be greater if more rows are employed. If spatial resolution of less than 7 µm is required, we will have to use different collection optics, with some possible sacrifice in monochromator throughput. The optics also limit us to observing a 2.5 mm segment of silk at one time, so if the conversion is not complete after drawing this far, we will have to observe static fibers to obtain spectra corresponding to endpoints in the conversion process.
|
|
Figure 5
Schematic representation of the Raman experiment, in this case observing the spinning of fibroin solution into fiber through an orifice.
Shown below is a representation of the ability to collect multiple spectra at points along the height of the CCD detector.
|
The sequence of spectroscopic tasks is approximately as follows:
1. Optimization of the Spectrum 1 system &endash; primarily choice of type of optical filter for rejection of Rayleigh scattering. Parts of this process may be complete before the start of funding.
2. Testing of ability to collect spectra using natural and artificial fibroin solutions, well-characterized fibroin forms such as heated and alcohol-precipitated fibroin, and synthetic oligopeptides with predictable secondary structures. While there are published reports of fibroin and silk Raman spectra, we must determine for ourselves that fluorescence levels are low enough in our samples to allow adequate S/N ratios for further secondary structural determination.
3. Alteration of sample illumination optics to allow illumination of vertical fiber from the side.
4. Design of a transparent artificial spinneret see above) for monitoring natural and biosynthetic fibroin solutions as they align in shear, but before they reach the air. A pulled quartz tube of dimensions similar to that of the silkworm duct would serve well.
5. Crude determination of secondary structure on static fibers by inspection of amide I and III bands, and comparison with our spectra from solutions, the published work of Magoshi et al. [14], and spectra of model compounds to be synthesized in our department. We will use molecular modeling techniques (see above) to interpret the vibrational spectra and determine conformations for the small model peptides.
6. Determination of secondary structure and water content in actively drawn fibers from spiders, silkworms, and biosynthetic fibroin from an artificial spinneret freestanding in air.
7. Accurate protein calibration using the Williams method.
8. Quantitative determination of secondary structure with the Williams method using previously collected data.
9. Introduction of fluorescent probe molecules into fibroin solutions to dynamically determine ionic strength, [Ca++], pH, and development of domains. Candidate probes include prodan as a probe of the polarity of the environment, and other dyes available from Molecular Probes for the ionic species.
10. Complete polarization studies &endash; these will require redirection of the laser beam into a backscattering direction using a small mirror in front of the collection lens to direct the beam toward the fiber along the monochromator axis.
The proposed interdisciplinary study will promote interaction between the fields of physical chemistry, materials processing, molecular biology and biological materials. While the liquid crystalline properties of some simple polypeptides have been noted previously, this has not yet been exploited to develop biological proteins as a source of high-performance engineering materials. Providing industrial quantities of such materials could constitute a new area of application for the emerging technology of protein synthesis through genetic engineering. The possibility of obtaining high strength, stiffness and toughness via processing aqueous solutions at ambient temperatures is certainly attractive.
The use of Raman equipment as described in this proposal allows the correlation of fiber characteristics with fiber drawing parameters (fibroin concentration, solvent conditions, orifice size, draw rate, temperature, relative humidity, etc.). Applied in the context of a fiber spinning line, this could be used to develop a process in which not only the composition but also the microstructure of the product is under continuous real-time feedback control.
It is worth noting that the majority of silk research is underway in countries traditionally associated with silk production. The progression of that research is towards mimicking the processing conditions that exist in nature. The proposed work recognizes the advantages of being at the leading edge of a technology that will provide high performance polymers from an essentially renewable, non-petroleum source at a low energy cost.
1. Shear, W. A., J. M. Palmer, et al. (1989). "A Devonian Spinneret: Early Evidence of Spiders and Silk Use." Science 246: 479-481.
2. Zemlin, J. C. (1968). "A study of the mechanical behavior of spider silks." Natick MA, U.S. Army Natick Laboratories. Report # 69-29-CM (AD 684333).
3. Gosline, J. M., M. E. DeMont, et al. (1986). "The structure and properties of spider silk." Endeavour 10(1): 37-43.
4. Kaplan, D. L., S. J. Lombardi, et al. (1991, in press). "Silks: Chemistry, Properties and Genetics." In: Biomaterials: Novel Materials from Biological Sources; D. Byrom (Ed.). New York, Stockton Press. 3-53.
5. Billmeyer, F. W. (1984). Textbook of Polymer Science. New York, John Wiley & Sons.
6. Lombardi, S. J. and D. L. Kaplan (1990). "Isolation, cloning, and physicochemical characterization of spider silk from the golder orb-weaver, Nephila clavipes." Polym. Preprints 31(1): 195-196.
7. Stinson, S. C. (1990). "Biotechnology providing springboard to new functional materials." C&EN (July 16): 26-32.
8. Yamaura, K., Y. Okumura, et al. (1985). "Flow-induced crystallinity of Bombyx Mori silk fibroin from regenerated aqueous solution and spinnability of its solution." J. Appl. Polym. Sci: Appl. Polym. Symp. 41: 205-220.
9. Xu, M. and R. V. Lewis (1990). "Structure of a protein superfiber: Spider dragline silk." Proc. Natl. Acad. Sci. USA 87(Sept.): 7120-7124.
10. Magoshi, J., M. Mizuide, et al. (1979). "Physical properties and structure of silk. VI. Conformational changes in silk fibroin induced by immersion in water at 2 to 130°C." J. Polym. Sci.: Polym. Phys. Ed. 17: 515-520.
11. Gosline, J. M., M. W. Denny, et al. (1984). "Spider silk as rubber." Nature 309(7): 551-552.
12. Iizuka, E. (1985). "Silk thread: mechanism of spinning and its mechanical properties." J. Appl. Polym. Sci.: Appl Polym. Symp. 41: 173-185.
13. Magoshi, J., Y. Magoshi, et al. (1985). "Crystallization, liquid crystal, and fiber formation of silk fibroin." J. Appl. Polym. Sci: Appl. Polym. Symp. 41: 187-204.
14. Magoshi, J., Y. Magoshi, et al. (1985). "Mechanism of fibre formation from liquid silk of silkworm Bombyx mori." Polymer. Commun. 26(Oct.): 309-311.
15. Zheng, S., G. Li, et al. (1989). "Raman spectroscopic investigation of the denaturation process of silk fibroin." Appl. Spect. 43(7): 1269-1272.
16. Weiss, R. A. and C. K. Ober, Ed. (1990). Liquid-Crystalline Polymers. American Chemical Society Symposium Series. Washington DC, American Chemical Society.
17. Kerkam, K., C. Viney, et al. (1991). "Liquid crystallinity of natural silk secretions." Nature 349: 596-598.
18. Viney, C. (1992, in press). "The Nature and Role of Liquid Crystalline Order in Silk Secretions." In: Structure, Cellular Synthesis and Assembly of Biopolymers; S. T. Case (Ed.). Heidelberg, Springer Verlag.
19. Iizuka, E. (1985). "Silk: an overview." Journal of Applied Polymer Science: Applied Polymer Symposia 41: 163-171.
20. Dobb, M. G. and J. E. McIntyre (1984). "Properties and Applications of Liquid-Crystalline Main-Chain Polymers." Advances in Polymer Science 60/61: 61-98.
21. Kerkam, K., D. L. Kaplan, et al. (1991). "Liquid Crystalline Characteristics of Natural Silk Secretions." In: Materials Synthesis Based on Biological Processes; M. Alper et al. (Eds). Pittsburgh, Materials Research Society; pp 239-244.
22. Asakura, T. (1986). "Structure of Bombyx mori silk fibroin in aqueous solution." Makromoleculare Chemie, Rapid Communications 7(12): 755-759.
23. Donald, A. M., C. Viney, et al. (1983). "Banded structures in oriented thermotropic polymers." Polymer 24: 155-159.
24. Thapar, H. and M. Bevis (1983). "The micromorphology of an injection moulded thermotropic liquid crystal polymer." Journal of Materials Science Letters 2: 733-736.
25. Zachariades, A. E., P. Navard, et al. (1984). "Deformation Studies of Liquid Crystalline Polymers." Molecular Crystals and Liquid Crystals 110: 93-107.
26. Chen, S., Y. Jin, et al. (1987). "Fibrillar structure in the oriented films of a thermotropic aromatic polyester." Polymer Communications 28: 208-211.
27. Fried, F. and P. Sixou (1988). "'Bands' and 'Torsads' Textures in Films and Threads of Hydroxypropyl Cellulose." Molecular Crystals and Liquid Crystals 158B: 163-184.
28. Ernst, B. and P. Navard (1989). "Band Textures in Mesomorphic (Hydroxypropyl) cellulose Solutions." Macromolecules 22: 1419-1422.
29. Putnam, W. S. and C. Viney (1991). "Effect of Processing Variables on Banded Textures in Hydroxypropyl Cellulose Solutions." Molecular Crystals and Liquid Crystals 199: 198-195.
30. Spiro, T. G. and B. P. Gaber (1977). "Laser Raman scattering as a probe of protein structure." Ann. Rev. Biochem. 46: 553-572.
31. Rath, P., O. Bousche, et al. (1991). "Fourier transform infrared evidence for a predominantly alpha-helical structure of the membrane bound channel forming COOH-terminal peptide of colicin E1." Biophys. J. 59: 516-22.
32. Archibald, D. D. (1990). "Structural studies of high aspect-ratio self-assembled lipid microstructures with the use of microscopy and FT-NIR-Raman spectroscopy." PhD thesis, University of Washington.
33. Jackson, M., P. I. Haris, et al. (1989). "Conformational transitions in poly(L-lysine): studies using Fourier transform infrared spectroscopy." Biochim. Biophys. Acta. 998: 75-79.
34. Dousseau, F. and M. Pézolet (1990). "Determination of the secondary structure content of proteins in aqueous solutions from their amide I and amide II infrared bands. Comparison between classical and partial least-squares methods." Biochemistry 29: 8771-9.
35. Lee, D. C., P. I. Haris, et al. (1990). "Determination of protein secondary structure using factor analysis of infrared spectra." Biochemistry 29: 9185-93.
36. Sarver, R. W. and W. C. Krueger (1991). "Protein secondary structure from Fourier transform infrared spectroscopy: a data base analysis." Anal. Biochem. 194: 89-100.
37. Williams, R. W. (1983). "Estimation of protein secondary structure from the laser Raman amide I spectrum." J. Mol. Biol. 166: 581-603.
38. Williams, R. W. (1986). "Protein secondary structural analysis using Raman amide I and amide III spectra." Meth. Enzym. 130: 311-331.
39. Bussian, B. M. and C. Sander (1989). "How to determine protein secondary structure in solution by Raman spectroscopy: Practical guide and test case DNase I." Biochemistry 28: 4271-7.
40. Yager, P., E. L. Chang, et al. (1984). "The secondary structure of acetylcholine receptor reconstituted in a single lipid component as determined by Raman spectroscopy." Biophys. J. 45: 26-28.
41. Putnam, W. S. and C. Viney (1990). "Observing the Relaxation of Molecular Orientation in Sheared Liquid Crystalline Polymer Solutions." In: Electron Microscopy 1990: XIIth International Congress for Electron Microscopy, Seattle WA, San Francisco Press; vol.4, pp 1092-1093.
42. Wilson, R. S. (1962). "The control of dragline spinning in the garden spider." Quart. J. Micr. Sci. 104(4): 557-571.
43. Flory, P. J. (1984). "Molecular Theory of Liquid Crystals." Advances in Polymer Science 59: 1-36.
44. Buseck, P. R., J. M. Cowley, et al., Ed. (1988). High-Resolution Transmission Electron Microscopy and Associated Techniques. New York, Oxford University Press.
45. Alexander, L. E. (1969). X-ray Diffraction Methods in Polymer Science. New York, John Wiley & Sons.
|
|
Return to Yager's Home Page Return to PY Research Project Page |