The Laboratory of Trisha Davis
Investigating the mechanisms of chromosome dynamics
Investigating the mechanisms of chromosome dynamics
Chromosome capture and movement during mitosis
The ultimate goal of mitotic spindle assembly is to arrange each chromosome with its sister chromatids attached to opposite poles via microtubule fibers. This ensures that when anaphase occurs, the two sister chromatids are pulled apart and partitioned one into each daughter cell. The assembly process is done by trial and error. Incorrect attachments are detected and detached to allow another attempt at correct attachment. Classic experiments have shown that tension is a key aspect of the error detection mechanism. The tension is sufficient to stretch the chromatin but not enough to shred the DNA or pull apart the spindle. How is tension established and maintained at the right level? How do the kinetochores and the motors work together to balance the forces in the spindle?
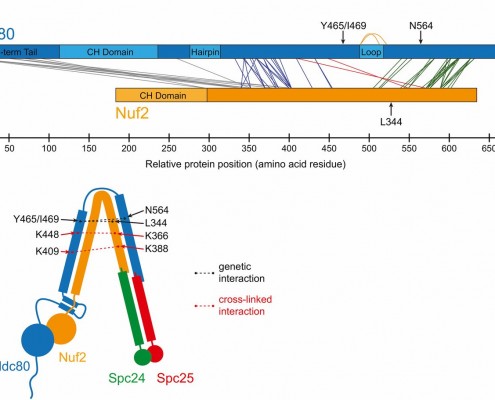
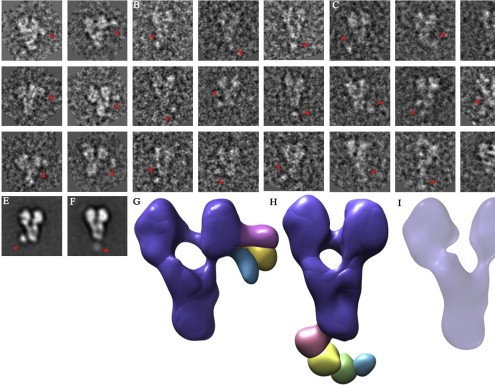
The kinetochore is a machine with a difficult task. It attaches the chromsomes to the dynamic microtubule fibers and must do so against ~20 pN of force, much more than is required to drag a chromosome through the cellular milieu. All chromosome segregation depends on this connection. The checkpoints are tuned to check for this connection and the tension it generates. We are using a wide range of approaches, including genetic analyses, proteomics, quantitative microscopy, biochemical assays, and computational modeling, to study the kinetochore as a molecular machine.
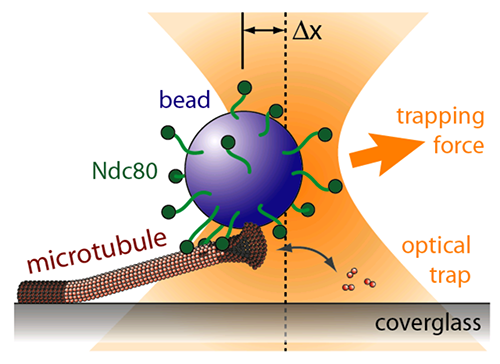
TIRF microscopy movie of human Ndc80 complex (green) stabilizing a depolymerizing microtubule (red). From: (Proc Natl Acad Sci U S A. 2012 Oct 2;109(40):16113-8. doi: 10.1073/pnas.1209615109. Epub 2012 Aug 20.)
Microtubule nucleation and organization during mitosis
Errors in chromosome segregation lead to aneuploidy, which results in birth defects, cancer or cell death. Accurate chromosome segregation is performed by a large molecular machine called the mitotic spindle. The mitotic spindle comprises many smaller machines including the centrosome, the microtubules themselves, the kinetochores where the microtubules attach to the chromosome and a multitude of microtubule motors.
In model organism Saccharomyces cerevisiae (budding yeast or baker’s yeast), the functional equivalent of the centrosome is a cylindrical multilayered structure called the spindle pole body (SPB). As the name suggests, the two SPBs form the poles of the mitotic spindle. They also organize the cytoplasmic microtubules that reach to the cortex of the cell and position the nucleus. Although SPBs do not resemble centrosomes on the ultrastructural level, many known SPB components have homologues found in the animal cell centrosome. The centrosome and the SPB perform similar functions and contain similar components. Thus, a detailed understanding of the regulation, structure and assembly of the yeast SPB will yield important insights into animal cell centrosomes. We are exploiting our ability to manipulate yeast genes at will to define the properties of this central and essential organizer. We are applying what we learn in yeast to an analysis of the centrosomal and chromosomal abnormalities that occur in human carcinomas.
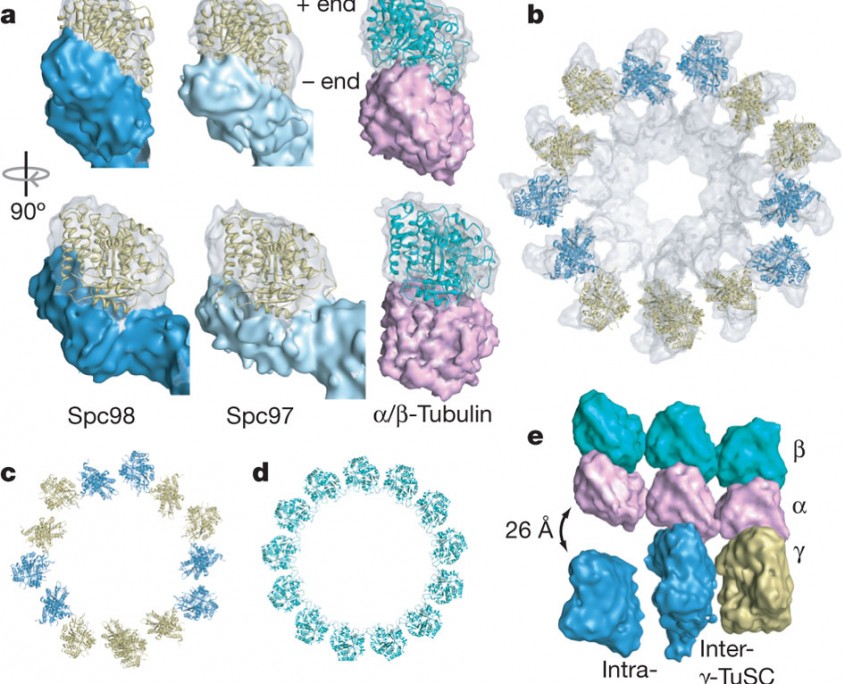
a, The electron microscopic densities of γ-tubulin (transparent) and the Spc97/Spc98-binding domains (opaque) are shown with the γ-tubulin crystal structure fitted in the density. The density is tilted relative to the helical axis so that in each case the plus end is vertical. An α/β-tubulin heterodimer is shown with simulated electron microscopic density for comparison. b, Thirteen γ-tubulins fit into one turn of the filament. Neighbouring γ-tubulins in the same γ-TuSC are the same colour. c, d, The γ-tubulin symmetry (c) is similar to that of a 13-protofilament microtubule (d), but separated intra-γ-TuSC γ-tubulins alternate with contacting inter-γ-TuSC γ-tubulins. e, To illustrate the mismatch between geometries, laterally interacting α/β-tubulin heterodimers were aligned to the filament so that the central α/β-tubulin makes longitudinal contacts to γ-tubulin. (Kollman et al. Nature. 2010 Aug 12;466(7308):879-82)