Differential Interaction of R-Mexiletine with the Local
Anesthetic Receptor Site on Cloned Brain and Heart Sodium Channel a
Subunits
Thomas Weiser, Yusheng Qu, William A. Catterall and
Todd Scheuer
Dept. of Pharmacology, University of Washington School of Medicine,
Seattle, WA 98195-7280, U.S.A. (T.W., Y.Q., W.A.C., T.S.) and Department
of CNS Research, Boehringer Ingelheim Pharma KG, D-55216, Ingelheim, Germany
(T.W.)
ABSTRACT
Mexiletine is a class 1 antiarrhythmic drug with neuroprotective effects
in models of brain ischemia due to inhibition of brain sodium channels.
We compared effects of R-mexiletine on wild-type (WT) and mutant rat brain
(rbIIA) and heart (rh1) sodium channel alpha subunits transiently expressed
in tsA-201 cells. R-mexiletine induced tonic and frequency-dependent block
of both brain and heart channels. The affinity of resting channels was
determined as the limiting minimum affinity reached at strongly negative
holding potentials (-140 mV or 160 mV). R-mexiletine bound with a 25-fold
(brain) or 35-fold (heart) higher affinity to depolarized channels. Affinities
for both resting and depolarized channels for R-mexiletine block were approximately
two-fold higher for heart than for brain channels. Mutations in transmembrane
segment IVS6 of heart (rhF1762A) and brain (rbF1764A and rbY1771A) channels,
which reduce block by other local anesthetics, also reduced block of inactivated
channels and use-dependent block by R-mexiletine. Unlike previous local
anesthetics studied, the strongest effect was observed for mutation rbY1771A.
A comparison of mutations of the homologous phenylalanine residue in brain
and heart channels showed striking differences in the effects of the mutations.
rbF1764A reduced drug block by drastically slowing R-mexiletine inhibition
of depolarized channels. rh1762A reduced R-mexiletine block by increasing
the rate of unbinding from depolarized or resting channels. Thus, rbF1764/rhF1762
is a critical determinant of local anesthetic block in both brain and heart
sodium channels, but its role differs in the two channel isoforms.
Introduction
Inhibitors of voltage-gated sodium channels are widely used clinically.
Blockers of cardiac sodium channels are potent antiarrhythmics, whereas
the inhibition of neuronal sodium channels is useful for local anesthesia
and treatment of epilepsy (Butterworth and Strichartz, 1990; Caron and
Libersa, 1997; Hondeghem and Katzung, 1984; Catterall, 1987; Ragsdale et
al., 1991). Moreover, inherited myotonias and cardiac arrhythmias caused
by mutations in skeletal muscle and cardiac sodium channels (Mitrovi'c
et al., 1995) can be effectively treated by class I antiarrhythmics (De
Luca et al., 1995). Thus, different sodium channel subtypes expressed in
neurons, cardiac or skeletal muscle cells can be modulated by molecules
of similar chemical structures. These overlapping actions bear risks and
benefits. On the one hand, local anesthetics inadvertently injected into
a blood vessel can cause severe cardiac arrhythmias. On the other hand,
some cardiac antiarrhythmics, including mexiletine, also penetrate the
blood-brain barrier, and have interesting neuroprotective properties (e.g.
Stys and Lesiuk, 1996).
Voltage-gated sodium channels are heteromultimeric proteins consisting
of a principal a subunit of 360 kD, as well
as a b1 subunit of 36 kD and, in the brain,
a b2 subunit of 33 kD. The a
subunit consists of four homologous transmembrane domains (I-IV), each
containing six transmembrane a-helical segments,
termed S1 through S6 (Catterall, 1992; Fozzard and Hanck, 1996). The principal
electrophysiological functions are mediated by the a
subunit; the b subunits have only minor effects
when the channels are heterologously expressed in mammalian cells (Isom
et al., 1995). The rat brain type IIA sodium channel a
subunit is a principal isoform expressed in the brain (Gordon et al., 1987;
Beckh et al., 1989) and its heterologous expression in mammalian cells
yields sodium currents with physiological and pharmacological properties
that are similar to those observed in rat brain neurons (West et al., 1992;
Ragsdale et al., 1991). The rH1 sodium channel a
subunit is the primary isoform expressed in the heart (Rogart et al., 1989;
Kallen et al., 1990) and expression of this isoform in Xenopus oocytes
or mammalian cells yields channels with physiological and pharmacological
properties characteristic of heart sodium channels (Cribbs et al., 1990;
White et al., 1991; Qu et al., 1994; Qu et al., 1995).
A study in which alanines were substituted for each amino acid in transmembrane
segment IVS6 identified mutations of two amino acid residues in the rbIIA
sodium channel, F1764A and Y1771A, that reduced block by the local anesthetic
etidocaine (Ragsdale et al., 1994). Mutation of these residues also reduced
block by a range of local anesthetic, anticonvulsant and anti-arrhythmic
compounds to varying degrees and with different rank order potencies, depending
on the structure of the particular blocker in rbIIA neuronal sodium channels
(Ragsdale et al., 1996) and m 1 skeletal muscle
channels (Wright et al., 1998; Wang et al., 1998). Mutation of the residue
homologous to rbIIA F1764 in the rH1 sodium channel, F1762, to ala also
resulted in loss of block of the rH1 channel by the quaternary lidocaine
analogue, QX314 in the rH1 (Qu et al., 1995). That study showed that rH1
F1762/rbIIA F1764 was a critical residue for local anesthetic block in
both channel backgrounds.
In the experiments described here, we compared the effects of R-mexiletine
on cloned rat brain and heart sodium channel a subunits
heterologously expressed in a common cellular background. We also examined
the effects of F1764A and Y1771A mutations in the rbIIA channel isoform
and compared the effects of mutation of phe rhF1762/rbF1764 in both heart
and brain backgrounds. We show that heart channels have an intrinsically
higher affinity for block by R-mexiletine than do brain channels when expressed
in the same cellular background. In contrast, lidocaine has a similar intrinsic
affinity for skeletal muscle and heart muscle sodium channels (Wright et
al., 1997). Mutations rbF1764A and rbY1771A both potently reduced R-mexiletine
affinity but with different specificity than for other local anesthetics.
Surprisingly, although mutations of the homolous phe residue in brain and
heart channels (rhF1762A/rbF1764A) both reduce block by R-mexiletine, the
kinetic details of their effects on are strikingly different. Thus, although
this phenylalanine is essential for local anesthetic block of both heart
and brain sodium channel isoforms, its role in that block is fundamentally
different in the two channel backgrounds. This detailed comparison of effects
of mutations of a single homologous amino acid in different channel backgrounds
yields novel insights into the role of this residue in heart and brain
channels.
Material and Methods
Cell maintenance and transient transfection for electrophysiological
recording. tsA-201 cells which are a subclone of HEK293 cells expressing
SV40 t-antigen, were a kind gift of Dr. Robert Dubridge (Cell Genesys,
Foster City, CA). Cells were maintained in DMEM/F12 media (Gibco/Life Technologies)
supplemented with 10% fetal bovine serum (Hyclone), 25 units/ml penicillin
and 25 m g/ml streptomycin (Sigma). An EcoRV
fragment containing mutants F1764A and Y1771A of RIIA in pVA2580 (Ragsdale
et al., 1996) was subcloned into pCDM8 containing the remainder of the
rIIA sodium channel a subunit as described (Linford
et al., 1998). rH1 (Rogart et al., 1989) and rH1 mutant F1762A in pCDM8
were described previously (Qu et al., 1996). tsA-201 cells were transiently
transfected with WT or mutant a subunits and
a vector encoding the human CD8 cell surface protein (EBO-pCD-leu2; American
Type Culture, Rockville, MD) for cell recognition as described (Margolskee
et al., 1993). Successfully transfected cells were labeled to recognize
them for recording using anti-CD8-coated polystyrene microspheres (Dynabeads
M-450 CD8, Dynal, Great Neck, NY) as described (Jurman et al., 1994).
Electrophysiological recording. Sodium currents were recorded
from transiently transfected tsA-201 cells in the whole cell voltage clamp
configuration (Hamill et al., 1981) at 22o C as described (Qu
et al., 1996). The extracellular solution contained (in mM): 140 NaCl,
5 CsCl, 1.8 CaCl2, 1.0 MgCl2, 10 glucose, and 10
HEPES (pH = 7.4, adjusted with NaOH). The intracellular solution contained
90 CsF, 50 CsCl, 10 CsEGTA, 10 NaF, 2 MgCl2 and 10 HEPES (pH=7.4
adjusted with CsOH). Recording pipettes had resistances of 0.8 to 1.8 MOhms
when filled with intracellular solution. The cells were bathed with the
effluent of a gravity-driven "sewer-pipe" perfusion system consisting of
a series of parallel tubes with each tube containing either control solution
or a solution containing R-mexiletine. Mexiletine is a racemate of S(+)-
and R(-) enantiomers, which have differential effects on sodium channels
(De Luca et al., 1995). In this study only the (-) enantiomer of R-mexiletine
was used. The compound was synthesized at the Department of Pharmaceutical
Chemistry at Boehringer Ingelheim KG (Ingelheim, Germany). R-mexiletine
was dissolved in extracellular solution at the highest concentration to
be used in an experiment and diluted with extracellular solution to each
of the other concentrations used. Solutions were changed by translating
the array of tubes so that the tube containing the appropriate concentration
was bathing the cell. The entire petri dish was continuously perfused with
control extracellular solution. Solution changes were complete within 2
s. Currents were recorded using an Axopatch 200B patch clamp amplifier
(Axon Instruments, Foster City, CA). Voltage clamp commands were delivered
and currents recorded using PClamp 6 controlling a Digidata 1200 interface
(Axon Instruments). Whole cell capacitance was compensated using the internal
voltage clamp circuitry and approximately 80% of series resistance was
compensated. Residual linear leakage and capacitance were subtracted using
a P/4 protocol when appropriate (Bezanilla and Armstrong, 1977). Data analysis
and curve fitting were performed using Sigma Plot (SPSS, Chicago) or Prism
(Graph-Pad Software, San Diego). All data points are the means of 3 to
6 experiments and grouped data are reported as ± SD.
Unless otherwise indicated, holding and test potentials were -100 and
0 mV for rbRIIA WT and F1764A, -120 and 0 mV for Y1771A, and -120 and -20
mV for rH1 heart sodium channels.
Results
Mutations F1764A and F1771A inhibit tonic and phasic block of brain
Na+ channels by R-mexiletine. The principal characteristics
of block by R-mexiletine are illustrated in Fig. 1A.
tsA-201 cells expressing rbIIA sodium channel a
subunits were voltage clamped at a holding potential of -100 mV, and the
control current trace was recorded in response to a 10 ms depolarization
to 0 mV. The cell was then exposed to R-mexiletine (100 mM)
in the absence of pulses. This was followed by a 5 Hz train of 100 pulses
(10 ms duration) to 0 mV. The first and one hundredth pulse of the train
are shown. Approximately 20% of the current was tonically blocked by this
concentration of R-mexiletine. The pulse train resulted in approximately
45% block in the steady state at this concentration and pulse frequency.
Thus, repetitive depolarization causes greater sensitivity to block by
R-mexiletine, as is observed for many local anesthetic compounds (Hille,
1977; Hondeghem and Katzung, 1984). Concentration-response curves were
obtained from similar experiments with IC50 values of 305 mM
and 165 mM for tonic and phasic block, respectively
(Fig. 1B). Thus, block increased approximately
2-fold during a 5 Hz train of this type.
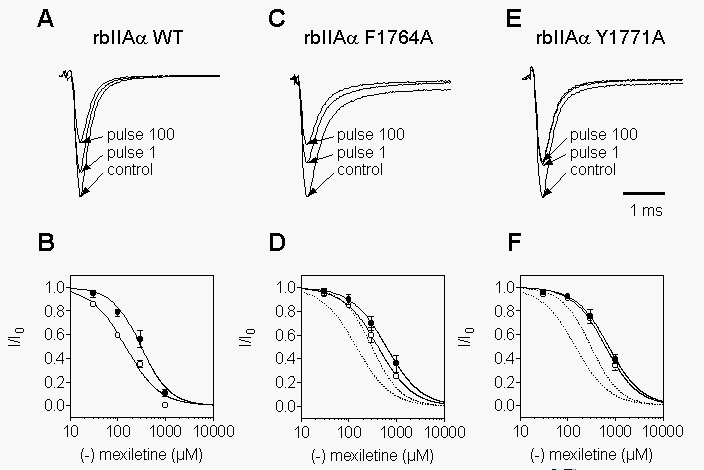 Two
mutants in transmembrane segment IVS6 of the rat brain sodium channel
a
subunit, F1764A and Y1771A, diminish block by diverse local anesthetic,
antiarrhythmic, and antiepileptic compounds and are proposed to contribute
to their binding sites to varying degrees (Ragsdale et al., 1994; Ragsdale
et al., 1996). Therefore, we have examined R-mexiletine block of these
mutant channels, as illustrated for 300 mM R-mexiletine
in Figs. 1C,E. Even at this 3-fold higher drug
concentration compared to WT (Fig. 1A), phasic
block during the train is reduced for F1764A and virtually abolished for
Y1771A. This is reflected in the IC50 values of 610 µM and 410
mM
for tonic and phasic block, respectively, of F1764A (Fig.
1D), and of 717 µM and 616
mM for
Y1771A (Fig. 1F). Thus, in comparison to WT, tonic
block is disrupted by 2-fold and 2.4-fold in mutants F1764A and Y1771A,
respectively. Similarly, phasic block measured at 5 Hz is reduced by 2.5-fold
and 3.7-fold in F1764A and Y1771A, respectively.
Two
mutants in transmembrane segment IVS6 of the rat brain sodium channel
a
subunit, F1764A and Y1771A, diminish block by diverse local anesthetic,
antiarrhythmic, and antiepileptic compounds and are proposed to contribute
to their binding sites to varying degrees (Ragsdale et al., 1994; Ragsdale
et al., 1996). Therefore, we have examined R-mexiletine block of these
mutant channels, as illustrated for 300 mM R-mexiletine
in Figs. 1C,E. Even at this 3-fold higher drug
concentration compared to WT (Fig. 1A), phasic
block during the train is reduced for F1764A and virtually abolished for
Y1771A. This is reflected in the IC50 values of 610 µM and 410
mM
for tonic and phasic block, respectively, of F1764A (Fig.
1D), and of 717 µM and 616
mM for
Y1771A (Fig. 1F). Thus, in comparison to WT, tonic
block is disrupted by 2-fold and 2.4-fold in mutants F1764A and Y1771A,
respectively. Similarly, phasic block measured at 5 Hz is reduced by 2.5-fold
and 3.7-fold in F1764A and Y1771A, respectively.
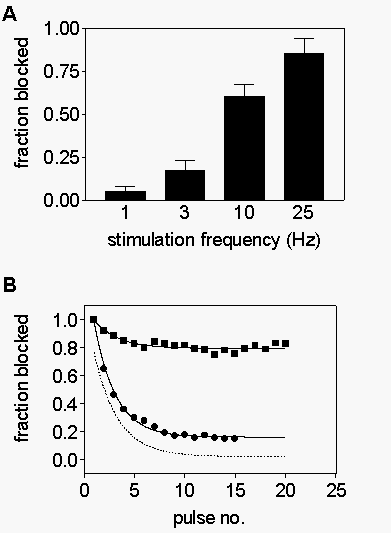
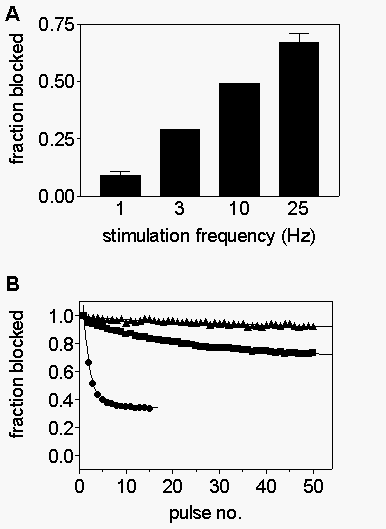 The
degree of phasic block becomes greater as stimulation frequency is increased
from 1 to 25 Hz (Fig. 2A), indicating that increased
frequency of depolarization strongly favors drug block. Fig.
2B shows examples of R-mexiletine block of WT (closed circles), F1764A
(squares), and Y1771A (triangles) channels during 25 Hz trains of depolarizations.
Whereas almost 70% of the current is blocked in the WT channels, only 27%is
blocked in F1764A and block develops much more slowly during the train.
Even at this frequency, phasic block of Y1771A is virtually nonexistent.
Use-dependent block results from preferential block of depolarized channels
and is strongly influenced by the rate of block during each depolarization
and the rate of unblock during intervening repolarizations. In order to
understand the mechanism by which phasic block is disrupted in F1764A and
Y1771A, each of these factors was examined.
The
degree of phasic block becomes greater as stimulation frequency is increased
from 1 to 25 Hz (Fig. 2A), indicating that increased
frequency of depolarization strongly favors drug block. Fig.
2B shows examples of R-mexiletine block of WT (closed circles), F1764A
(squares), and Y1771A (triangles) channels during 25 Hz trains of depolarizations.
Whereas almost 70% of the current is blocked in the WT channels, only 27%is
blocked in F1764A and block develops much more slowly during the train.
Even at this frequency, phasic block of Y1771A is virtually nonexistent.
Use-dependent block results from preferential block of depolarized channels
and is strongly influenced by the rate of block during each depolarization
and the rate of unblock during intervening repolarizations. In order to
understand the mechanism by which phasic block is disrupted in F1764A and
Y1771A, each of these factors was examined.

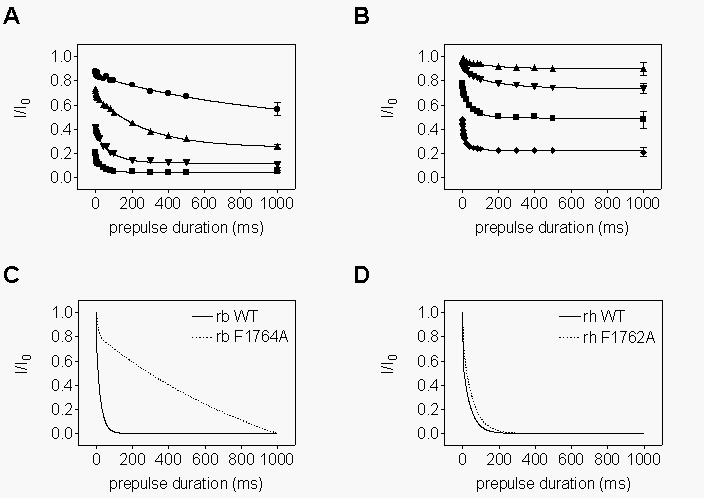
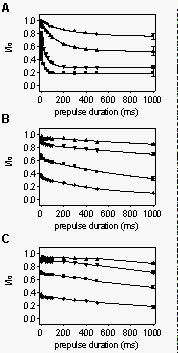 Disruption
of voltage-dependent block in mutants F1764A and Y1771A. We first investigated
the kinetics of R-mexiletine binding to depolarized sodium channels. To
measure the time course of binding to depolarized channels, prepulses to
0 mV of variable duration (0.5 - 1000 ms) were applied in the absence or
presence of increasing concentrations of R-mexiletine followed by a test
pulse. Prepulse and test pulses were separated by an interval of 45 ms
at the holding potential of -100 mV. This interval was long enough to allow
unblocked channels to recover completely from inactivation (unpublished
results), but short enough so that R-mexiletine unbinding was minimal (<10%,
see Fig. 7A). Data values in the presence of the compound were normalized
to the controls. In WT, as well as F1764A channels, the data points could
be best fitted by double-exponential functions (Fig.
3A,B). Block of Y1771A mutant channels was so slow that relatively
little block developed, even after 1 s of depolarization (Fig.
3C). The small magnitude of the decay prevented fitting it reproducibly.
The slow, weak block of Y1771A is consistent with the lack of phasic block
(see Fig. 2B).
Disruption
of voltage-dependent block in mutants F1764A and Y1771A. We first investigated
the kinetics of R-mexiletine binding to depolarized sodium channels. To
measure the time course of binding to depolarized channels, prepulses to
0 mV of variable duration (0.5 - 1000 ms) were applied in the absence or
presence of increasing concentrations of R-mexiletine followed by a test
pulse. Prepulse and test pulses were separated by an interval of 45 ms
at the holding potential of -100 mV. This interval was long enough to allow
unblocked channels to recover completely from inactivation (unpublished
results), but short enough so that R-mexiletine unbinding was minimal (<10%,
see Fig. 7A). Data values in the presence of the compound were normalized
to the controls. In WT, as well as F1764A channels, the data points could
be best fitted by double-exponential functions (Fig.
3A,B). Block of Y1771A mutant channels was so slow that relatively
little block developed, even after 1 s of depolarization (Fig.
3C). The small magnitude of the decay prevented fitting it reproducibly.
The slow, weak block of Y1771A is consistent with the lack of phasic block
(see Fig. 2B).
As most channels are inactivated for depolarizations longer than 5 ms
to 0 mV, the slow time constant of block for WT and F1764A can be attributed
to R-mexiletine binding to inactivated channels. The most likely interpretation
of the faster component is that it reflects rapid binding of R-mexiletine
via the open sodium channel. Consistent with this idea, the time constant
and magnitude of the exponential component represented by the fast time
constant were also concentration-dependent. They corresponded roughly to
the period of time over which channels opened. However, the exact time
course and magnitude of this component was not resolved precisely enough
in these experiments to determine its nature in detail. Like the slow component,
its magnitude was greatly reduced in the mutant channels (Fig.
3B,C).
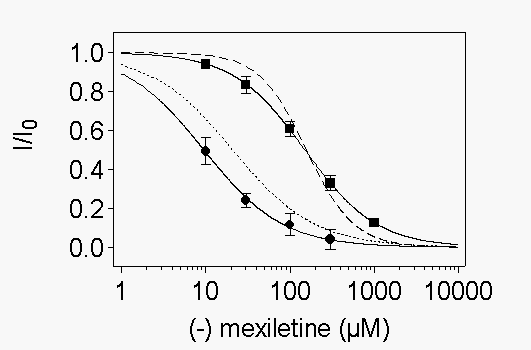
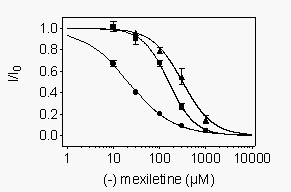 We
also examined drug block at potentials where most channels are inactivated
but activation is minimal. WT and mutant channels were depolarized to -40
mV (-60 mV for Y1771A) for 1 s to allow drug to bind. At this potential
more than 98% of channels were inactivated, thus allowing us to study the
effect of R-mexiletine on inactivated channels. The membrane potential
was then returned to the holding potential of 100 mV (-120 mV for Y1771A)
for 10 ms followed by a 10 ms test pulse to 0 mV. A more negative potential
was used for Y1771A to compensate for its more negative voltage dependence
of inactivation when expressed in tsA-201 cells (Weiser and Scheuer, unpublished
observations). In control experiments virtually all of the channels that
were inactivated during the 40 mV prepulse recovered from inactivation
during the 10 ms repolarization period, whereas only 2% of the R-mexiletine-blocked
channels recovered from drug block (see Fig. 7A
below). Concentration-response curves obtained using this protocol showed
that R-mexiletine inhibited WT channels with an IC50 of 20.7 µM (Fig.
4). Inactivated mutant channels had reduced affinity for block by R-mexiletine
with IC50 values of 156 mM and 309 mM
for F1764A and Y1771A channels, respectively (Fig.
4). Thus, the mutation F1764A disrupted block of the inactivated state
by 7.5 fold, and the mutation Y1771A caused a 14.9-fold disruption of inactivated
state block. For WT channels, the ratio between the tonic block affinity
and this measurement of affinity for inactivated channels was 14.7. In
contrast, for mutant F1764A this ratio was only 3.9 and for mutant Y1771A
the ratio was 2.3. Thus, these mutations affect R-mexiletine affinity for
inactivated channels more than they affect affinity for resting channels.
We
also examined drug block at potentials where most channels are inactivated
but activation is minimal. WT and mutant channels were depolarized to -40
mV (-60 mV for Y1771A) for 1 s to allow drug to bind. At this potential
more than 98% of channels were inactivated, thus allowing us to study the
effect of R-mexiletine on inactivated channels. The membrane potential
was then returned to the holding potential of 100 mV (-120 mV for Y1771A)
for 10 ms followed by a 10 ms test pulse to 0 mV. A more negative potential
was used for Y1771A to compensate for its more negative voltage dependence
of inactivation when expressed in tsA-201 cells (Weiser and Scheuer, unpublished
observations). In control experiments virtually all of the channels that
were inactivated during the 40 mV prepulse recovered from inactivation
during the 10 ms repolarization period, whereas only 2% of the R-mexiletine-blocked
channels recovered from drug block (see Fig. 7A
below). Concentration-response curves obtained using this protocol showed
that R-mexiletine inhibited WT channels with an IC50 of 20.7 µM (Fig.
4). Inactivated mutant channels had reduced affinity for block by R-mexiletine
with IC50 values of 156 mM and 309 mM
for F1764A and Y1771A channels, respectively (Fig.
4). Thus, the mutation F1764A disrupted block of the inactivated state
by 7.5 fold, and the mutation Y1771A caused a 14.9-fold disruption of inactivated
state block. For WT channels, the ratio between the tonic block affinity
and this measurement of affinity for inactivated channels was 14.7. In
contrast, for mutant F1764A this ratio was only 3.9 and for mutant Y1771A
the ratio was 2.3. Thus, these mutations affect R-mexiletine affinity for
inactivated channels more than they affect affinity for resting channels.
Compounds that bind preferentially to depolarized and inactivated channels
generally shift the voltage-dependence of steady-state inactivation. This
is illustrated in 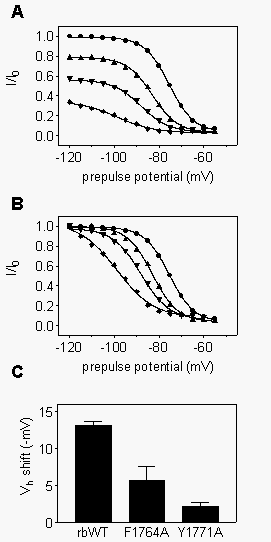 Fig.
5 for R-mexiletine block of WT channels. We applied test pulses to
0 mV after 1s prepulses to different potentials in the absence or presence
of various concentrations of R-mexiletine (Fig. 5A).
To demonstrate the concentration-dependent shift in inactivation curves,
the same data were normalized to the maximum value obtained with the Boltzmann
fits (Fig. 5B). For these WT channels, increasing
concentrations of mexiletine progressively shifted the steady-state inactivation
curves to more negative potentials, consistent with R-mexiletine preferently
binding to the inactivated state of the channel as measured directly above.
Since preferential binding to inactivated channels was reduced in mutants
F1764A and Y1771A, inactivation curves would be expected to be less strongly
shifted as a function of drug concentration. Consistent with this expectation,
steady-state inactivation curves were shifted less in the presence of 300
m
M R-mexiletine in comparison to WT (Fig. 5C).
Similar results were observed at other R-mexiletine concentrations (Table
1).
Fig.
5 for R-mexiletine block of WT channels. We applied test pulses to
0 mV after 1s prepulses to different potentials in the absence or presence
of various concentrations of R-mexiletine (Fig. 5A).
To demonstrate the concentration-dependent shift in inactivation curves,
the same data were normalized to the maximum value obtained with the Boltzmann
fits (Fig. 5B). For these WT channels, increasing
concentrations of mexiletine progressively shifted the steady-state inactivation
curves to more negative potentials, consistent with R-mexiletine preferently
binding to the inactivated state of the channel as measured directly above.
Since preferential binding to inactivated channels was reduced in mutants
F1764A and Y1771A, inactivation curves would be expected to be less strongly
shifted as a function of drug concentration. Consistent with this expectation,
steady-state inactivation curves were shifted less in the presence of 300
m
M R-mexiletine in comparison to WT (Fig. 5C).
Similar results were observed at other R-mexiletine concentrations (Table
1).
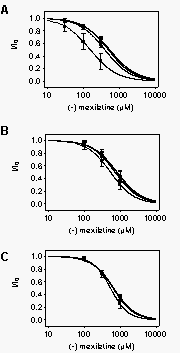 Affinity
of resting WT and mutant rat brain Na+ channels for
R-mexiletine. The previous experiments show that R-mexiletine binds
with high affinity to inactivated brain Na+ channels.
R-mexiletine also induced tonic inhibition of sodium currents with lower
apparent affinity (see Fig. 1). This reduced affinity
could result from a high affinity for the small number of inactivated sodium
channels at the holding potential of -100 mV. Tonic block that is entirely
due to stabilization of residual inactivated channels should continue to
decrease with further hyperpolarization as that inactivation is reversed.
Therefore, we asked whether increased holding potential hyperpolarization
reduces tonic block by R-mexiletine (Fig. 6).
Cells were voltage clamped for 20 s to 80 mV (squares), -100 mV
(triangles), -120 mV (inverted triangles), and -140 mV (diamonds)
for WT (Fig. 6A) and F1764A (Fig.
6B), or 100 mV (squares), -120 mV (triangles), -140
mV (inverted triangles), and -160 mV (diamonds) for Y1771A (Fig.
6C), prior to a test pulse to 0 mV. Concentration-response curves for
block by R-mexiletine were determined at each holding potential. For each
construct, the curves for holding potentials more negative than -120 mV
superimpose, indicating that R-mexiletine affinity approaches a limiting
value with hyperpolarization that indicates the IC50 for resting channels.
The limiting IC50 value for resting WT channels (533 µM at 140 mV,
Table 1) was less than for F1764A (813 µM) or Y1771A (655 µM)
channels, indicating that the affinity of resting channels for R-mexiletine
was reduced in the mutants. The dependence of the IC50 for R-mexiletine
block on the holding potential (compare diamonds with squares
in Fig. 6) was strongest in WT channels, less
pronounced for F1764A, and almost absent for Y1771A mutant channels.
Affinity
of resting WT and mutant rat brain Na+ channels for
R-mexiletine. The previous experiments show that R-mexiletine binds
with high affinity to inactivated brain Na+ channels.
R-mexiletine also induced tonic inhibition of sodium currents with lower
apparent affinity (see Fig. 1). This reduced affinity
could result from a high affinity for the small number of inactivated sodium
channels at the holding potential of -100 mV. Tonic block that is entirely
due to stabilization of residual inactivated channels should continue to
decrease with further hyperpolarization as that inactivation is reversed.
Therefore, we asked whether increased holding potential hyperpolarization
reduces tonic block by R-mexiletine (Fig. 6).
Cells were voltage clamped for 20 s to 80 mV (squares), -100 mV
(triangles), -120 mV (inverted triangles), and -140 mV (diamonds)
for WT (Fig. 6A) and F1764A (Fig.
6B), or 100 mV (squares), -120 mV (triangles), -140
mV (inverted triangles), and -160 mV (diamonds) for Y1771A (Fig.
6C), prior to a test pulse to 0 mV. Concentration-response curves for
block by R-mexiletine were determined at each holding potential. For each
construct, the curves for holding potentials more negative than -120 mV
superimpose, indicating that R-mexiletine affinity approaches a limiting
value with hyperpolarization that indicates the IC50 for resting channels.
The limiting IC50 value for resting WT channels (533 µM at 140 mV,
Table 1) was less than for F1764A (813 µM) or Y1771A (655 µM)
channels, indicating that the affinity of resting channels for R-mexiletine
was reduced in the mutants. The dependence of the IC50 for R-mexiletine
block on the holding potential (compare diamonds with squares
in Fig. 6) was strongest in WT channels, less
pronounced for F1764A, and almost absent for Y1771A mutant channels.
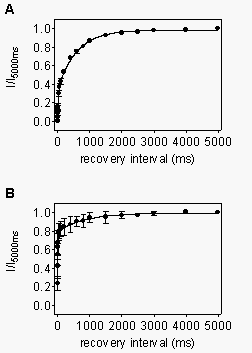 Recovery
from use-dependent block by R-mexiletine. In addition to increased
affinity for depolarized channels, the degree of use-dependent block during
a train of depolarizations is also determined by the rate of recovery from
such block between the depolarizations. Therefore, we analyzed the kinetics
of recovery from use-dependent block by R-mexiletine. Use-dependent inhibition
was induced by a conditioning train of 10 depolarizing pulses at 10 Hz
frequency in the presence of 300 µM R-mexiletine. The recovery was
characterized by applying test pulses after recovery intervals of variable
duration (1-5000 ms). The data obtained by this procedure could be fitted
with the sum of two exponential functions, with the time-constant of the
faster component describing the recovery from inactivation of unblocked
channels, and the slower time constant, the recovery of the mexiletine-blocked
fraction of channels. For WT channels (Fig. 7A),
the slow time-constant due to drug unbinding was 571 ms, indicating that
mexiletine left its binding sites relatively rapidly. For F1764A channels
(Fig. 7B), the total amount of use-dependent block
induced by the train was reduced, in comparison to WT. However, the time-constant
of drug unbinding was even slightly slower than in the WT channels (858
ms). Little phasic block of Y1771A mutant channels could be developed,
even at high stimulation frequencies (Fig. 2B).
Therefore, recovery was not analyzed for this mutant. These results indicate
that the reduced use-dependent block in mutant F1764A is caused by reduced
drug binding during each depolarization and not from accelerated unbinding
between depolarizations.
Recovery
from use-dependent block by R-mexiletine. In addition to increased
affinity for depolarized channels, the degree of use-dependent block during
a train of depolarizations is also determined by the rate of recovery from
such block between the depolarizations. Therefore, we analyzed the kinetics
of recovery from use-dependent block by R-mexiletine. Use-dependent inhibition
was induced by a conditioning train of 10 depolarizing pulses at 10 Hz
frequency in the presence of 300 µM R-mexiletine. The recovery was
characterized by applying test pulses after recovery intervals of variable
duration (1-5000 ms). The data obtained by this procedure could be fitted
with the sum of two exponential functions, with the time-constant of the
faster component describing the recovery from inactivation of unblocked
channels, and the slower time constant, the recovery of the mexiletine-blocked
fraction of channels. For WT channels (Fig. 7A),
the slow time-constant due to drug unbinding was 571 ms, indicating that
mexiletine left its binding sites relatively rapidly. For F1764A channels
(Fig. 7B), the total amount of use-dependent block
induced by the train was reduced, in comparison to WT. However, the time-constant
of drug unbinding was even slightly slower than in the WT channels (858
ms). Little phasic block of Y1771A mutant channels could be developed,
even at high stimulation frequencies (Fig. 2B).
Therefore, recovery was not analyzed for this mutant. These results indicate
that the reduced use-dependent block in mutant F1764A is caused by reduced
drug binding during each depolarization and not from accelerated unbinding
between depolarizations.
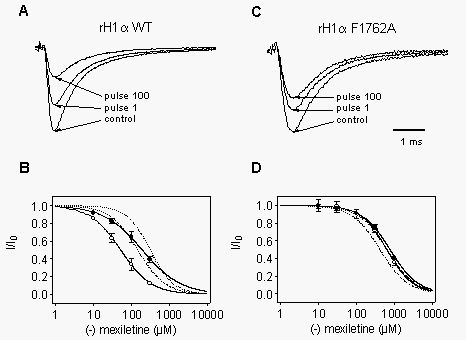 Block
of WT and F1762A mutant rat heart sodium channels by R-mexiletine.
Block of sodium channels formed by expression of rat heart a
subunits by R-mexiletine was also studied and compared to brain channels.
Tonic and phasic block were initially examined during a train of 100 depolarizations
using a protocol analogous to that of Fig. 1 (Fig.
8A). For these experiments, the holding and test potentials were 20
mV negative to those used for brain to account for the more negative activation
and inactivation properties of the rat heart channel (Fozzard and Hanck,
1996; Weiser and Scheuer, unpublished observations). Concentration-response
curves (Fig. 8B) obtained using this protocol
gave IC50 values of of 165 and 52 µM for tonic (closed circles) and
phasic (open circles) block, respectively. These values were lower than
those for WT brain channels (Fig. 1A, C; dotted
lines in Fig. 8B), suggesting a higher affinity
of R-mexiletine for heart sodium channels.
Block
of WT and F1762A mutant rat heart sodium channels by R-mexiletine.
Block of sodium channels formed by expression of rat heart a
subunits by R-mexiletine was also studied and compared to brain channels.
Tonic and phasic block were initially examined during a train of 100 depolarizations
using a protocol analogous to that of Fig. 1 (Fig.
8A). For these experiments, the holding and test potentials were 20
mV negative to those used for brain to account for the more negative activation
and inactivation properties of the rat heart channel (Fozzard and Hanck,
1996; Weiser and Scheuer, unpublished observations). Concentration-response
curves (Fig. 8B) obtained using this protocol
gave IC50 values of of 165 and 52 µM for tonic (closed circles) and
phasic (open circles) block, respectively. These values were lower than
those for WT brain channels (Fig. 1A, C; dotted
lines in Fig. 8B), suggesting a higher affinity
of R-mexiletine for heart sodium channels.
We also examined the F1762A mutant of the heart channel, which is homologous
to the brain F1764A mutation (Qu et al., 1995). As with its brain counterpart,
tonic (closed circles) and phasic (open circles) inhibition by R-mexiletine
were reduced for this mutant (Fig. 8C) with IC50
values of 748 µM and 619 µM, respectively (Fig.
8D). In contrast to the WT channels, the affinity for the mutant heart
channel measured in this way was actually lower than for its F1764A brain
counterpart (dotted lines in Fig. 8D).
 As
with brain channels, the degree of phasic block increased with stimulation
frequency from 5% at 1 Hz to 85% at 25 Hz. However, for the equivalent
concentration and frequency, greater block was reached for heart than for
brain sodium channels (compare Fig. 9A, heart,
with Fig. 2A, brain). This is consistent with
the higher sensitivity of heart channels to phasic block by R-mexiletine.
As
with brain channels, the degree of phasic block increased with stimulation
frequency from 5% at 1 Hz to 85% at 25 Hz. However, for the equivalent
concentration and frequency, greater block was reached for heart than for
brain sodium channels (compare Fig. 9A, heart,
with Fig. 2A, brain). This is consistent with
the higher sensitivity of heart channels to phasic block by R-mexiletine.
Phasic inhibition (Fig. 9B) of mutant F1762A
(squares) was much less pronounced than for the WT heart channel (circles).
A 25 Hz stimulus train in the presence of 100 µM mexiletine induced
only about 20% inhibition in the F1762A mutant compared to 85% in the WT.
Despite the large disruption by this mutant, the rates of development of
use-dependent block were quite similar for rH WT and F1762 (compare the
dashed line in Fig. 9B which is the fit to
the F1762A data scaled to be similar in magnitude to the WT data). For
the 100 µM R-mexiletine concentration shown, the stimulation-dependent
decline of peak current responses could be fitted with single exponential
functions with similar time constants for the WT and mutant channels (on-rate
WT: 0.39/pulse; on-rate for F1762A: 0.42/pulse). This contrasts dramatically
with the F1764A mutant brain channel where the development of phasic block
during a train in the mutant channel was dramatically slower than WT (Fig.
2B).
 Voltage-dependent
inhibition of WT and F1762A mutant heart sodium channels. The difference
in the effects of mutants on the kinetics of use-dependent block in the
brain and heart channels suggested that the mutants might have different
effects on the detailed kinetics of R-mexiletine action. To investigate
the onset of inhibition in more detail, we applied depolarizing prepulses
of various lengths prior to a test pulse, a protocol analogous to that
of Fig. 3. For a given concentration, the degree
of depolarization-dependent block was greater for the WT rH1 channel (Fig.
10A) in comparison to the rbIIA channel (Fig.
3A), consistent with the increased block of the heart channel during
trains of pulses presented in Fig. 9. As for brain
channels, data could be best fitted by double exponentials. Significant
binding to the heart channel occurs after most channels have inactivated,
and suggests that mexiletine can bind to inactivated channels. The onset
of block in response to depolarization in mutant F1762A showed that the
final level of block at the end of a 1s depolarization was greatly reduced
for a given concentration (Fig. 10B), but the
kinetics of the development of block were not dramatically affected. This
is demonstrated in Fig. 10D where the fits to
the onset data for rh1 WT and F1762A are normalized and superimposed, showing
their similar time courses. This similarity contrasts with the drastically
different time courses observed for rbIIA WT and mutant F1764A (Fig.
10C).
Voltage-dependent
inhibition of WT and F1762A mutant heart sodium channels. The difference
in the effects of mutants on the kinetics of use-dependent block in the
brain and heart channels suggested that the mutants might have different
effects on the detailed kinetics of R-mexiletine action. To investigate
the onset of inhibition in more detail, we applied depolarizing prepulses
of various lengths prior to a test pulse, a protocol analogous to that
of Fig. 3. For a given concentration, the degree
of depolarization-dependent block was greater for the WT rH1 channel (Fig.
10A) in comparison to the rbIIA channel (Fig.
3A), consistent with the increased block of the heart channel during
trains of pulses presented in Fig. 9. As for brain
channels, data could be best fitted by double exponentials. Significant
binding to the heart channel occurs after most channels have inactivated,
and suggests that mexiletine can bind to inactivated channels. The onset
of block in response to depolarization in mutant F1762A showed that the
final level of block at the end of a 1s depolarization was greatly reduced
for a given concentration (Fig. 10B), but the
kinetics of the development of block were not dramatically affected. This
is demonstrated in Fig. 10D where the fits to
the onset data for rh1 WT and F1762A are normalized and superimposed, showing
their similar time courses. This similarity contrasts with the drastically
different time courses observed for rbIIA WT and mutant F1764A (Fig.
10C).
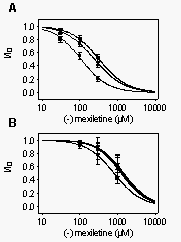
 As
for the brain channel, we recorded concentration-response curves after
depolarizations that inactivated most channels, but produced minimal activation.
WT- or F1762A-expressing cells were depolarized to -60 mV for 1 s followed
by a 10 ms return to the holding potential. The degree of inactivation
induced by the conditioning pulse was then assayed using a test pulse to
-20 mV. R-mexiletine inhibited WT channels at -60 mV with an IC50 of 9.4
µM, whereas 149 µM were necessary for half-maximum block of
F1762A channels (Fig. 11A). Thus, the IC50 values
for the inhibition of WT heart or brain channels differed by a factor of
about 2, whereas the concentrations for half-maximum block of the rH F1762A
and rbIIA F1764A were similar (156 versus 149 µM). The amount of
inactivation at the prepulse potential was greater than 97% for each channel
type. The similar amounts of inactivation for the different channels allows
a direct comparison of the IC50 values obtained with the prepulse experiments.
Thus, the affinity of R-mexiletine for depolarized heart WT channels is
about two-fold higher than for brain WT channels, and is disrupted by 7.2-
and 16.5-fold in rb F1764A and rh F1762A, respectively.
As
for the brain channel, we recorded concentration-response curves after
depolarizations that inactivated most channels, but produced minimal activation.
WT- or F1762A-expressing cells were depolarized to -60 mV for 1 s followed
by a 10 ms return to the holding potential. The degree of inactivation
induced by the conditioning pulse was then assayed using a test pulse to
-20 mV. R-mexiletine inhibited WT channels at -60 mV with an IC50 of 9.4
µM, whereas 149 µM were necessary for half-maximum block of
F1762A channels (Fig. 11A). Thus, the IC50 values
for the inhibition of WT heart or brain channels differed by a factor of
about 2, whereas the concentrations for half-maximum block of the rH F1762A
and rbIIA F1764A were similar (156 versus 149 µM). The amount of
inactivation at the prepulse potential was greater than 97% for each channel
type. The similar amounts of inactivation for the different channels allows
a direct comparison of the IC50 values obtained with the prepulse experiments.
Thus, the affinity of R-mexiletine for depolarized heart WT channels is
about two-fold higher than for brain WT channels, and is disrupted by 7.2-
and 16.5-fold in rb F1764A and rh F1762A, respectively.
The shift of steady-state inactivation curves towards more negative
potentials was also observed for R-mexiletine-blocked heart sodium channels
(Fig. 11B, circles). For a given concentration,
the effect was more pronounced for rhWT channels than for their rbWT counterparts
(see Table 1). As was the case for the rb F1764A mutant, the shifts were
reduced in the rh F1762A channels (Fig. 11B,
squares;
Fig. 11C). In contrast to WT channels, the shifts were comparable for rh
F1762A and rb F1764A mutants at the same R-mexiletine concentration (compare
Figs.
5C and 11C; Table 1).
 Affinity
of resting WT and mutant rat heart Na+ channels for R-mexiletine.
We
also analyzed the concentration dependence of tonic inhibition of heart
sodium channels at increasingly negative potentials. For rhWT channels,
tonic inhibition by R-mexiletine was similar at holding potentials of 140
mV and 160 mV (Fig. 12A;
inverted triangles,
diamonds). Fits to these curves gave an IC50 of 165 mM,
approximately twice the affinity of the rbWT channel. Higher affinities
were measured at holding potentials of 120 mV and 100 mV (Fig.
12A, triangles,
squares) as expected if the affinity
for inactivated channels is higher than for resting channels. The affinity
for resting channels was reduced in mutant rhF1762A at -140 mV and -160
mV(Fig. 12B, inverted triangles, diamonds),
and low affinity block was also observed at -120 mV (Fig.
12B, triangles), consistent with the reduced affinity of the
inactivated state. Fits of the curves at 140 and 160 mV yielded an IC50
of 748 mM, similar to the rbF1764A value of
614 mM. Thus, resting mutant rbF1764A brain
and rhF1762A heart channels had similar affinity for R-mexiletine, whereas
WT heart channels had an approximately 2-fold higher affinity (Table 1).
Affinity
of resting WT and mutant rat heart Na+ channels for R-mexiletine.
We
also analyzed the concentration dependence of tonic inhibition of heart
sodium channels at increasingly negative potentials. For rhWT channels,
tonic inhibition by R-mexiletine was similar at holding potentials of 140
mV and 160 mV (Fig. 12A;
inverted triangles,
diamonds). Fits to these curves gave an IC50 of 165 mM,
approximately twice the affinity of the rbWT channel. Higher affinities
were measured at holding potentials of 120 mV and 100 mV (Fig.
12A, triangles,
squares) as expected if the affinity
for inactivated channels is higher than for resting channels. The affinity
for resting channels was reduced in mutant rhF1762A at -140 mV and -160
mV(Fig. 12B, inverted triangles, diamonds),
and low affinity block was also observed at -120 mV (Fig.
12B, triangles), consistent with the reduced affinity of the
inactivated state. Fits of the curves at 140 and 160 mV yielded an IC50
of 748 mM, similar to the rbF1764A value of
614 mM. Thus, resting mutant rbF1764A brain
and rhF1762A heart channels had similar affinity for R-mexiletine, whereas
WT heart channels had an approximately 2-fold higher affinity (Table 1).
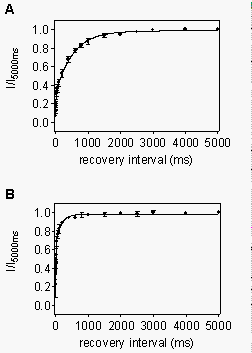 Recovery
of heart sodium channels from use-dependent block by R-mexiletine is accelerated
in mutant rhF1762A. Frequency-dependent block was induced by a train
of 10 conditioning pulses to -20 mV (10 Hz frequency). Recovery was assayed
by a test pulse to -20 mV after repolarizations to 120 mV of increasing
duration. Approximately 75% of rhWT channels were blocked by such a train
in the presence of 100 mM R-mexiletine, as indicated
by the fraction of channels recovering with the slower exponential slow
time course at the holding potential. For rhWT channels, the time constant
for this recovery was 529 ms (Fig. 13A). Frequency-dependent
block of rhF1762A channels in the presence of 300 mM
R-mexiletine produced only approximately 25% block using this protocol,
consistent with the reduced affinity of the depolarized channel in this
mutant (Fig. 9). Recovery of blocked channels
occurred with a time constant of 168 ms, 3-fold faster than the rhWT channel
(Fig. 13B). Thus, accelerated recovery from drug
block on repolarization in mutant F1762A must contribute substantially
to the reduced frequency-dependent block observed in this mutant.
Recovery
of heart sodium channels from use-dependent block by R-mexiletine is accelerated
in mutant rhF1762A. Frequency-dependent block was induced by a train
of 10 conditioning pulses to -20 mV (10 Hz frequency). Recovery was assayed
by a test pulse to -20 mV after repolarizations to 120 mV of increasing
duration. Approximately 75% of rhWT channels were blocked by such a train
in the presence of 100 mM R-mexiletine, as indicated
by the fraction of channels recovering with the slower exponential slow
time course at the holding potential. For rhWT channels, the time constant
for this recovery was 529 ms (Fig. 13A). Frequency-dependent
block of rhF1762A channels in the presence of 300 mM
R-mexiletine produced only approximately 25% block using this protocol,
consistent with the reduced affinity of the depolarized channel in this
mutant (Fig. 9). Recovery of blocked channels
occurred with a time constant of 168 ms, 3-fold faster than the rhWT channel
(Fig. 13B). Thus, accelerated recovery from drug
block on repolarization in mutant F1762A must contribute substantially
to the reduced frequency-dependent block observed in this mutant.
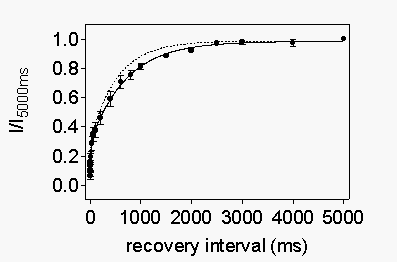 Channel-specific
amino acid residues in the IVS6 transmembrane segment of the rat heart
sodium channel slow recovery from use-dependent block by R-mexiletine.
The
quaternary local anesthetic QX314 blocks WT heart, but not brain, Na+
channels from the extracellular medium (Baumgarten et al., 1991). Mutations
in the extracellular portion of transmembrane segment IVS6 can provide
an extracellular access pathway in the brain channel (Ragsdale et al.,
1994). Conversely, chimeric mutants rhLTT-FVS and rhT1775V, which change
rat heart residues L1752, T1755 and T1756 to their brain counterparts,
occlude such a pathway in the heart channel (Qu et al., 1995). These mutants
that prevent extracellular access of local anesthetics also slow recovery
from use-dependent block by QX314 (Qu et al., 1995), presumably because
the drug molecule leaves its binding site more slowly via the extracellular
opening of the pore of the mutant channels. To examine whether this extracellular
access pathway is important for recovery from use-dependent block by R-mexiletine,
we compared the recovery of rhWT channels with mutant rhLTT-FVS and rhT1775V.
The rhLTT-FVS and rhT1755V mutants behaved comparably to the rhWT counterpart
in terms of their affinities for R-mexiletine block of resting and depolarized
channels (Table 1). The primary effect of these mutations was on recovery
from use-dependent block by R-mexiletine (Fig. 14).
For both rhLTT-FVS and rhT1755V, recovery from use-dependent block was
slightly, but significantly slower than for rhWT channels. The slow time
constant of recovery was 779 ms for rhLTT-FVS, 784 ms for rhT1755V (Fig.
14,
dotted line), and 529 ms for rhWT. These results show that
the difference in R-mexiletine block of brain and heart channels is not
caused by the differences in these amino acid residues which control extracellular
access. Differences in release from the local anesthetic receptor site
may be primarily responsible for the different rates of recovery.
Channel-specific
amino acid residues in the IVS6 transmembrane segment of the rat heart
sodium channel slow recovery from use-dependent block by R-mexiletine.
The
quaternary local anesthetic QX314 blocks WT heart, but not brain, Na+
channels from the extracellular medium (Baumgarten et al., 1991). Mutations
in the extracellular portion of transmembrane segment IVS6 can provide
an extracellular access pathway in the brain channel (Ragsdale et al.,
1994). Conversely, chimeric mutants rhLTT-FVS and rhT1775V, which change
rat heart residues L1752, T1755 and T1756 to their brain counterparts,
occlude such a pathway in the heart channel (Qu et al., 1995). These mutants
that prevent extracellular access of local anesthetics also slow recovery
from use-dependent block by QX314 (Qu et al., 1995), presumably because
the drug molecule leaves its binding site more slowly via the extracellular
opening of the pore of the mutant channels. To examine whether this extracellular
access pathway is important for recovery from use-dependent block by R-mexiletine,
we compared the recovery of rhWT channels with mutant rhLTT-FVS and rhT1775V.
The rhLTT-FVS and rhT1755V mutants behaved comparably to the rhWT counterpart
in terms of their affinities for R-mexiletine block of resting and depolarized
channels (Table 1). The primary effect of these mutations was on recovery
from use-dependent block by R-mexiletine (Fig. 14).
For both rhLTT-FVS and rhT1755V, recovery from use-dependent block was
slightly, but significantly slower than for rhWT channels. The slow time
constant of recovery was 779 ms for rhLTT-FVS, 784 ms for rhT1755V (Fig.
14,
dotted line), and 529 ms for rhWT. These results show that
the difference in R-mexiletine block of brain and heart channels is not
caused by the differences in these amino acid residues which control extracellular
access. Differences in release from the local anesthetic receptor site
may be primarily responsible for the different rates of recovery.
Discussion
R-Mexiletine block of native and heterologously expressed heart sodium
channels is similar. R-Mexiletine has been described as a fast-onset,
use-dependent blocker of heart sodium channels. Using rat ventricular myocytes,
Yatani and Akaike (1985) found an IC50 of 28 µM for tonic inhibition.
Those data were obtained using a holding potential of -80 mV, which caused
approximately 50% channel inactivation under their experimental conditions.
The time-constant for recovery from block at a holding potential of -90
mV was 370 ms. In the same preparation, Ono et al. (1994) reported half-maximum
inhibition of depolarized channels to be 15 µM. Heterologous expression
of WT human heart sodium channels resulted in steady-state block of depolarized
channels with an IC50 of 15 µM (Wang et al., 1997). These data are
consistent with the IC50 of 9.4 µM that we obtained for R-mexiletine
block of expressed rat heart sodium channels under depolarized conditions
in the present study. Thus, the data on rat heart sodium channel a
subunits presented here are in good agreement with those obtained previously.
Higher intrinsic affinity for block of heart versus brain sodium
channels. We examined the affinity of the resting and of the inactivated
states of heart and brain channels for R-mexiletine. The best estimate
of affinity for resting channels is to determine block of peak current
using increasingly negative holding potentials. Block by R-mexiletine reaches
a limiting affinity which is not reduced as the holding potential is made
more negative (Figs. 6 and 12;
Table 1). This limiting IC50 is 533 µM in the brain channel and 321
µM in the heart channel. Thus, when elicited from the fully resting
state, the heart sodium channel is approximately 1.6 times more sensitive
to block than the brain channel. Likewise, our best estimate for block
of depolarized channels obtained using a depolarizing prepulse (Figs.
4 and 11; Table 1) indicates an approximately
2-fold higher sensitivity of depolarized heart channels to block by R-mexiletine
than of brain channels. These findings contrast with a recent report comparing
lidocaine sensitivity for resting brain and heart sodium channels indicating
that there is little intrinsic difference in sensitivity of the isoforms
to block of resting channels by lidocaine (Wright et al., 1997). Evidently,
R-mexiletine does have approximately two-folder higher intrinsic affinity
for heart sodium channels. This preferential block of heart sodium channels
likely contributes to the therapeutic effect of mexiletine on cardiac arrhythmias.
Interactions with rbF1764/rHF1762 are responsible for the difference
in affinity for brain and heart sodium channels. The greater affinity
of heart sodium channels for block by R-mexiletine was largely abolished
by the mutations F1764A and F1762A. The affinity of the inactivated states
of these two mutants were not significantly different (KI=149
µM or 156 µM, respectively, Table 1), and IC50 values at different
potentials were also similar for these two mutants. These results suggest
that interactions with this key phenylalanine are primarily responsible
for the higher affinity of heart sodium channels for R-mexiletine.
Studies of effects of other local anesthetics on brain or skeletal muscle
isoforms of sodium channels have demonstrated that an additional stable
blocked state is present in the heart sodium channel that is absent in
brain and skeletal muscle isoforms (Zamponi et al., 1993; Gingrich et al.,
1993). If R-mexiletine block of the heart channel exhibits a similar stable
blocked state that is absent in the brain isoform, it could be this state
that is disrupted in the heart mutant F1762A.
Common mechanism of R-mexiletine block of sodium channels. Despite
the different affinities for brain and heart sodium channel a
subunits, the effects on WT channels of both subtypes are qualitatively
similar. In both channel types R-mexiletine caused use-dependent block,
shifted steady-state inactivation curves toward more negative potentials,
and had higher affinities for depolarized, inactivated channels in comparison
to hyperpolarized resting ones. Furthermore, onset of R-mexiletine block
during depolarization occurred in rapid and slower exponential phases representing
binding to open and inactivated states of both channel types. These observations
suggest that the basic mechanism of R-mexiletine block of brain and heart
sodium channels is similar.
The literature contains considerable discussion of whether mexiletine
binds to the open or inactivated state of depolarized channels (e.g. Ono
et al., 1995; Wang et al., 1997). Our data demonstrate a clear biexponential
onset of R-mexiletine block of depolarized channels. The most straightforward
explananation of such a time course is that the fast and slow components
of block represent R-mexiletine binding to open and inactivated channels,
respectively. The slower exponential phase of block of depolarized sodium
channels occurs largely at times when the vast majority of sodium channels
are inactivated. Clearly, R-mexiletine can bind to inactivated channels.
The time constant of the fast component was not well resolved in our experiments
but its time course overlaps the period when channels are open and and
its rate increases with increasing concentration. Furthermore, the fraction
of block attributable to the fast time constant increases with concentration
(Figs. 3 and 10; Table
1). This is expected if R-mexiletine can bind to either open or inactivated
states of depolarized channels but it can achieve that block more rapidly
if the channels are open.
Homologous mutations F1764A and F1762A affect R-mexiletine block
in different ways. In spite of the similarities in steady-state block
of the mutant brain and heart channels, the mutations at F1764/1762 disrupted
R-mexiletine block in strikingly different ways. Mutation F1764A of the
brain channel had a pronounced effect on the rate of association of the
drug with the depolarized channel (Figs. 3 and
10C).
After drug block had developed, there was little effect of the mutation
on drug unbinding from depolarized or hyperpolarized channels (Fig.
7). Thus, the major effects of replacing this phe with ala was to greatly
slow R-mexiletine binding to the mutant brain channel; once bound, the
stability of the complex was comparable to WT. Change of the analogous
amino acid in the heart sodium channel had quite a different effect. For
this mutant, the kinetics of drug association with the heart channel were
comparable to association with the WT channel (Fig.
10D). However, drug dissociation from the depolarized (Fig.
10B) and hyperpolarized mutant channel (Fig.
13) was much faster than from the WT channel. Thus, in the brain channel
the presence of the WT phe at position 1764 facilitates development of
the blocked state but has small effects on the dissociation of the complex.
In the heart sodium channel, the homologous phe at position 1762 stabilizes
the drug in its binding site and prevents unbinding. This is reflected
in a 3.5-fold increase in IC50 for binding to the resting F1762A mutant
channel at 160 mV, a decreased level of steady-state block at -20 mV (Fig.
10), a 15-fold increase in IC50 at -60 mV (Fig.
11A) in the mutant channel, and a 3-fold decrease in the time constant
for drug unbinding from channels after repolarization to 120 mV (Fig.
13). The greater effects on depolarized as opposed to hyperpolarized
channels indicate that the F1762A mutation preferentially disrupts R-mexiletine
binding to depolarized states of the channel.
Common and divergent effects of IVS6 mutations on block of brain
sodium channels by R-mexiletine and other local anesthetic/antiarrhythmic
compounds. Each point mutation in transmembrane segment IVS6 that was
known to reduce block by other local anesthetic, anticonvulsant and antiarrhythmic
drugs, also reduced use-dependent R-mexiletine block of rbIIA sodium channels
(Ragsdale et al., 1994; Qu et al., 1995). Use- and voltage-dependent block
of brain sodium channels by etidocaine is reduced in mutants F1764A and
Y1771A, and F1764 is more important for binding than Y1771 (Ragsdale et
al., 1994). Although these two mutations also reduced the affinity for
R-mexiletine, Y1771A reduced R-mexiletine block much more profoundly than
F1764A. Similarly, (Ragsdale et al., 1996) found that effects of a range
of sodium channel inhibitors on mutants F1764A and Y1771A were all reduced
compared to WT channels. However, rank orders of potencies for use-dependent
block were different for the tested compounds. Block by lidocaine was reduced
more by mutant F1764A than by mutant Y1771A, whereas, for flecainide, the
efficacy of the mutations was reversed. Thus, sodium channel modulators
of different chemical structure may interact in different ways with the
local anesthetic receptor site in segment IVS6 of the sodium channel.
Overall this study demonstrates that sodium channel blockers like mexiletine
can have different affinities for binding to the local anesthetic receptor
site brain and heart sodium channel subtypes. Mutations of equivalent amino
acids can have strongly different effects on the kinetics of channel inhibition
in different subtypes. These findings suggest different roles for equivalent
amino acids in the different subtypes and provide a rationale for the development
of tissue-specific sodium channel blockers with defined kinetic properties.
Acknowledgements
The major portion of this study was performed during a sabbatical leave
of T. Weiser from Boehringer Ingelheim. T.W. would like to express his
gratitude to Dr. N. Mayer, Prof. R. Hammer and Prof. B. Wetzel (Boehringer
Ingelheim) for offering him this opportunity and for critical discussions
of the work. The authors also thank Dr. M. Grauert for his assessment of
the purity of the enantiomer(s) of mexiletine and Ms. Elizabeth M. Sharp
for assistance with molecular biology.
References
Baumgarten CM, Makielski JC and Fozzard HA (1991) External site for
local anesthetic block of cardiac Na+ channels. J Mol Cell
Cardiol 23 (Suppl 1):85-93.
Beckh S, Noda M, Lubbert H and Numa S (1989) Differential regulation
of three sodium channel messenger RNAs in the rat central nervous system
during development. EMBO J 8:3611-3616.
Bezanilla F and Armstrong CM (1977) Inactivation of the sodium channel.
I. Sodium current experiments. J Gen Physiol 70:549-566.
Butterworth JF and Strichartz GR (1990) Molecular mechanisms of local
anesthesia: A review. Anesthesiology 72:711-734.
Caron J and Libersa C (1997) Adverse effects of class I antiarrhythmic
drugs. Drug Saf 17:8-36.
Catterall WA (1987) Common modes of drug action on Na+ channels:
Local anesthetics, antiarrhythmics and anticonvulsants. Trends Pharmacol
Sci 8:57-65.
Catterall WA (1992) Cellular and molecular biology of voltage-gated
sodium channels. Physiol Rev 72:S15-48.
Cribbs LL, Satin J, Fozzard HA and Rogart RB (1990) Functional expression
of the rat heart I Na+ channel isoform. Demonstration of properties
characteristic of native cardiac Na+ channels. FEBS Lett 275:195-200.
De Luca A, Natuzzi F, Lentini G, Franchini C, Tortorella V and Conte
CD (1995) Stereoselective effects of mexiletine enantiomers on sodium currents
and excitability characteristics of adult skeletal muscle fibers. Naunyn
Schmiedebergs Arch Pharmacol 352:
Fozzard HA and Hanck DA (1996) Structure and function of voltage-dependent
sodium channels: Comparison of brain II and cardiac isoforms. Physiol
Rev 76:887-926.
Gingrich KJ, Beardsley D and Yue DT (1993) Ultra-deep blockade of Na+
channels by a quaternary ammonium ion: catalysis by a transition-intermediate
state? J Physiol Lond 471:319-341.
Gordon D, Merrick D, Auld V, Dunn R, Goldin AL, Davidson N and Catterall
WA (1987) Tissue-specific expression of the RI and RII sodium channel subtypes.
Proc
Natl Acad Sci U S A 84:8682-8686.
Hamill OP, Marty A, Neher E, Sakmann B and Sigworth FJ (1981) Improved
patch-clamp techniques for high-resolution current recording from cells
and cell-free membrane patches. Pflugers Arch 391:85-100.
Hille B (1977) Local anesthetics: hydrophilic and hydrophobic pathways
for the drug-receptor reaction. J Gen Physiol 69:497-515.
Hondeghem LM and Katzung BG (1984) Antiarrhythmic agents: the modulated
receptor mechanism of action of sodium and calcium channel blocking drugs.
Annu
Rev Pharmacol Toxicol 24:387-423.
Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ and Catterall
WA (1995) Functional co-expression of the b1
and type IIA a subunits of sodium channels in
a mammalian cell line. J Biol Chem 270:3306-3312.
Jurman ME, Boland LM, Liu Y and Yellen G (1994) Visual identification
of individual transfected cells for electrophysiology using antibody-coated
beads. Biotechniques 17:876-881.
Kallen RG, Sheng ZH, Yang J, Chen LQ, Rogart RB and Barchi RL (1990)
Primary structure and expression of a sodium channel characteristic of
denervated and immature rat skeletal muscle. Neuron 4:233-242.
Linford NJ, Cantrell AR, Qu Y, Scheuer T and Catterall WA (1998) Interaction
of batrachatoxin with the local anesthetic receptor site in transmembrane
segment IVS6 of the voltage-gated sodium channel. Proc Natl Acad Sci
U S A 95:13947-13952.
Margolskee RF, McHendry-Rinde B and Horn R (1993) Panning transfected
cells for electrophysiological studies. Biotechniques 15:906-911.
Mitrovi'c N, George AL, Jr., Lerche H, Wagner S, Fahlke C and Lehmann
Horn F (1995) Different effects on gating of three myotonia-causing mutations
in the inactivation gate of the human muscle sodium channel. J Physiol
Lond 487:107-114.
Ono M, Sunami A and Hiraoka M (1995) Interaction between external Na+
and mexiletine on Na+ channel in guinea-pig ventricular myocytes.
Pflugers
Arch 431:101-109.
Ono M, Sunami A, Sawanobori T and Hiraoka M (1994) External pH modifies
sodium channel block by mexiletine in guinea pig ventricular myocytes.
Cardiovasc
Res 28:973-979.
Qu Y, Isom LL, Westenbroek RE, Rogers JC, Tanada TN, McCormick KA, Scheuer
T and Catterall WA (1995) Modulation of cardiac Na+ channel
expression in Xenopus oocytes by b1 subunits.
J
Biol Chem 270:25696-25701.
Qu Y, Rogers J, Tanada T, Scheuer T and Catterall WA (1994) Modulation
of cardiac Na+ channels expressed in a mammalian cell line and
in ventricular myocytes by protein kinase C. Proc Natl Acad Sci U S
A 91:3289-3293.
Qu Y, Rogers J, Tanada T, Scheuer T and Catterall WA (1995) Molecular
determinants of drug access to the receptor site for antiarrhythmic drugs
in the cardiac Na+ channel. Proc Natl Acad Sci U S A92:11839-11843.
Qu YS, Rogers JC, Tanada TN, Catterall WA and Scheuer T (1996) Phosphorylation
of S1505 in the cardiac Na+ channel inactivation gate is required
for modulation by protein kinase C. J Gen Physiol 108:375-379.
Ragsdale DS, McPhee JC, Scheuer T and Catterall WA (1994) Molecular
determinants of state-dependent block of Na+ channels by local
anesthetics. Science 265 :1724-1728.
Ragsdale DS, McPhee JC, Scheuer T and Catterall WA (1996) Common molecular
determinants of local anesthetic, antiarrhythmic, and anticonvulsant block
of voltage-gated Na+ channels. Proc Natl Acad Sci U S A93:9270-9275.
Ragsdale DS, Scheuer T and Catterall WA (1991) Frequency and voltage-dependent
inhibition of type IIA Na+ channels, expressed in a mammalian
cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs.
Mol
Pharmacol 40:756-765.
Rogart RB, Cribbs LL, Muglia LK, Kephart DD and Kaiser MW (1989) Molecular
cloning of a putative tetrodotoxin-resistant rat heart Na+ channel
isoform. Proc Natl Acad Sci U S A 86:8170-8174.
Stys PK and Lesiuk H (1996) Correlation between electrophysiological
effects of mexiletine and ischemic protection in central nervous system
white matter. Neuroscience 71:27-36.
Wang DW, Yazawa K, Makita N, George AL and Bennett PB (1997) Pharmacological
targeting of long-QT mutant sodium channels. J Clin Invest 99:1714-1720.
Wang GK, Quan C and Wang S (1998) A common local anesthetic receptor
for benzocaine and etidocaine in voltage-gated m1
Na+ channels. Pflugers Arch 435:293-302.
West JW, Scheuer T, Maechler L and Catterall WA (1992) Efficient expression
of rat brain type IIA Na+ channel a
subunits in a somatic cell line. Neuron 8:59-70.
White MM, Chen LQ, Kleinfield R, Kallen RG and Barchi RL (1991) SkM2,
a Na+ channel cDNA clone from denervated skeletal muscle, encodes
a tetrodotoxin-insensitive Na+ channel. Mol Pharmacol39:604-608.
Wright SN, Wang S-Y, Kallen RG and Wang GK (1997) Differences in steady-state
inactivation between Na channel isoforms affect local anesthetic binding
affinity. Biophys J 73:779-788.
Wright SN, Wang SY and Wang GK (1998) Lysine point mutations in Na+
channel D4-S6 reduce inactivated channel block by local anesthetics. Mol
Pharmacol 54 :733-739.
Yatani A and Akaike N (1985) Blockade of sodium current in isolated
single cells from rat ventricle with mexiletine and disopyramide. J
Mol Cell Cardiol 17:467-476.
Zamponi GW, Doyle DD and French RJ (1993) State-dependent block underlies
the tissue specificity of lidocaine action on batrachotoxin-activated cardiac
sodium channels. Biophys J 65:91-100.
FOOTNOTES
These experiments were supported by NIH Program Project Grant P01 HL44948
(W.A.C., Principal Investigator).
TABLE 1
Block of WT and mutant brain and heart channels by R-mexiletine.
| Channel Type |
rb WT |
rb F1764A |
rb Y1771A |
rh WT |
rh F1762A |
| IC50 Tonic block (µM)a |
305
|
614
|
718
|
165
|
748
|
| Phasic block (µM)a |
148
|
410
|
617
|
52
|
618
|
| Tau recovery (ms)b |
571
|
858
|
n.d.
|
529
|
168
|
| h¥
shift (mV)c |
|
|
|
|
|
|
30 µM
|
n.d.
|
n.d.
|
n.d.
|
-3.2
|
n.d.
|
|
100 µM
|
-7.5
|
-2.0
|
n.d.
|
-9.0
|
n.d.
|
|
300 µM
|
-13.1
|
-5.8
|
-2.2
|
-17.3
|
-6.2
|
|
1000 µM
|
-20.9
|
-11.6
|
-8.3
|
n.d.
|
-12.4
|
| IC50 Tonic block (µM)d |
|
|
|
|
|
|
-80 mV
|
155
|
531
|
n.d.
|
n.d.
|
n.d.
|
|
-100 mV
|
394
|
714
|
532
|
107
|
806
|
|
-120 mV
|
498
|
803
|
629
|
245
|
1261
|
|
-140 mV
|
533
|
813
|
655
|
321
|
1468
|
| Tau onset of block (ms)e |
|
|
|
|
|
|
10 µM
|
326
|
n.d.
|
n.d.
|
878
|
n.d.
|
|
30 µM
|
152
|
2475
|
n.d.
|
266
|
128
|
|
100 µM
|
52
|
2598
|
n.d.
|
85
|
187
|
|
300 µM
|
24
|
1139
|
n.d.
|
44
|
56
|
|
1000 µM
|
n.d.
|
549
|
n.d.
|
n.d.
|
65
|
| IC50 depolarized block (µM)f |
20.7
|
156
|
309
|
9.4
|
149
|
-
Tonic and phasic block measured as described in the legend to Fig. 1 for
brain channels and Fig. 8 for heart channels.
-
Time constant for recovery from block induced by a train of 10 depolarizations
to 0 mV as described in the legend to Fig. 7 for brain channels and Fig.
13 for heart channels.
-
Shift in the half inactivation voltage in the presence of the indicated
R-mexiletine concentration determined as described in the legends to Figs.
5 and 11.
-
Tonic block determined at different holding potentials as described in
the legends to Fig. 6 and 12.
-
Time constant for onset of block at 0 mV determined as described in the
legends to Figs. 3 and 10.
-
IC50 for steady-state block at a holding potential of 40 mV (brain) or
60 mV (heart).
FIGURE LEGENDS
Fig. 1. Mutations in the IVS6 region of brain type sodium channel
a
subunits differently influence tonic and phasic block by mexiletine. A
single pulse to 0 mV was applied in the absence of drug. Then R-mexiletine
was washed into the bath for 30 s to 3 min without pulsing. Control experiments
showed that equilibrium R-mexiletene block was reached within seconds.
Finally, a 5 Hz train of 100 depolarizations (10 ms duration) to 0 mV was
applied to voltage-clamped tsA-201 cells that had been transfected with
rat brain sodium channel a subunits. A,C,E,
Current traces evoked by the single pulse in drug-free solution (control)
and by pulse 1 and pulse 100 of the train in the presence of 100 µM
(A) or 300 µM R-mexiletine (C and E). The inhibition in response
to pulse 1 is referred to as tonic block, and to pulse 100 is referred
to as phasic block. B,D,F, Concentration-response curves for tonic (closed
circles) and phasic (open circles) block of rbWT (B), rb F1764A (D) and
rbY1771A (F). The solid lines are fits to the data for each construct with
IC50 values of 305 and 148 µM for tonic and phasic block of rbWT
channels, respectively, (B), 614 and 410 µM for rbF1764A (D), and
718 and 617 µM for Y1771A (F). Dotted lines in D and F represent
the fitted curves for rbWT channels taken from B.
Fig. 2. Frequency-dependent inhibition of rhWT and mutant sodium
channels. A, Trains of 15 pulses to 0 mV (10 ms duration) were applied
in the presence of 300 µM mexiletine. The fraction of current blocked
by trains of different frequencies was determined as [1-(peak current evoked
by pulse 15/ peak current evoked by pulse 1)]. B, Trains of 25 Hz depolarizations
were applied to cells expressing rbWT(circles), rbF1764A(squares), and
rbY1771A(triangles) channels. Peak current normalized to peak current in
pulse 1 is plotted versus pulse number.
Fig. 3. Onset of mexiletine block during depolarizations. Onset
of R-mexiletine block of sodium currents in cells expressing rbIIA WT (A),
F1764A (B), and Y1771A (C) sodium channels. Currents were elicited by test
pulses to 0 mV that had been preceded by conditioning pulses to 0 mV of
variable duration (0-1000 ms) to develop drug block followed by a 45 ms
return to the holding potential of 100 mV (-120 mV for Y1771A) to allow
drug-free channels to recover from inactivation. Peak amplitudes in the
presence of R-mexiletine were normalized to those determined with the same
protocol in control. Double-exponential functions were fit to the data
points. The time constants of the fast components were assumed to reflect
the rate of inhibition via the open channels, whereas the slow components
describes the rate of block of inactivated channels. The block of rbY1771A
channels was too small to be fit accurately, and the data points were connected
by straight lines. Concentrations of R-mexiletine were 10 µM (circles),
30 µM (triangles), 100 µM (inverted triangles), 300 µM
(squares), and 1000 µM (diamonds), respectively. Note that the range
of concentrations was different for rbWT and mutant channels.
Fig 4. Mutants rbF1764A and rbY1771A reduce block of depolarized
sodium channels. The inhibition of inactivated sodium channels was investigated
using a 1 s depolarizing prepulse to -40 mV (-60 mV for rbY1771A) to inactivate
most channels and allow R-mexiletine to bind to the inactivated channels.
The membrane potential was then returned to the holding potential of -100
mV (-120 mV for Y1771A) for 10 ms to allow recovery of drug-free channels
to recover from inactivation. A test pulse to 0 mV was then applied. R-mexiletine
block of peak test pulse current is plotted as a function of R-mexiletine
concentration for WT (circles), rbF1764A (squares), and rbY1771A (triangles)
channels. The solid lines are fits of a logistic equation to the data with
IC50 values of 20.7 µM (WT), 156 mM (rbF1764A)
and 309 mM (rbY1771A).
Fig. 5. Hyperpolarizing shifts of steady-state availability curves
in response to R-mexiletine. Steady-state availability was investigated
using 1 s prepulses to a variable potential followed by a test pulse to
0 mV. A, Response of a typical cell in control (circles) and in the presence
of 100 µM (triangles), 300 µM (inverted triangles) and 1000
µM (diamonds) R-mexiletine normalized to the largest currents in
control. The solid lines are fits of a Boltzmann relationship to the data
points. B, Each curve from (A) has been normalized to its largest value
to emphasize the concentration-dependent shift in voltage dependence. C,
The shift of inactivation curves is reduced in mutants rbF1764A and rbY1771A,
compared to rbWT channels. The bar graph shows the shift of the midpoints
of inactivation curves in response to 300 µM R-mexiletine for the
three rIIA constructs.
Fig. 6. Limiting potency of R-mexiletine at negative holding
potentials. Concentration-response curves for block by R-mexiletine were
determined using different holding potentials for block of rbWT (A), rbF1764A
(B) and rbY1771A (C). For A and B the holding potentials tested were -80
mV (squares), -100 mV (triangles), -120 mV (inverted triangles), and 140
mV (diamonds). The potentials in (C) were -100 mV (squares), -120 mV (triangles),
-140 mV (inverted triangls), and 160 mV (diamonds). For all constructs,
block saturated at more hyperpolarized potentials.
Fig. 7. Recovery from use-dependent block for rbWT and rbF1764A
mutant channels. Use-dependent inhibition by 300 µM mexiletine was
generated by applying a 10 Hz conditioning train of 10 pulses to 0 mV.
The membrane potential was then returned to 100 mV and a test pulse to
0 mV was applied after a recovery interval of variable duration. The rates
of recovery at 100 mV for WT (A) and mutant rbF1764A (B) were generated
by plotting the peak test pulse current normalized to its value after 5000
ms recovery as a function of recovery interval. The data were fitted by
double-exponential functions, where the faster time constant reflected
the recovery from inactivation of unblocked channels, and the slower, the
recovery from block by mexiletine. The time constants for recovery from
use-dependent block by R-mexiletine were 571 and 858 ms for rbWT and rbF1764A
channels respectively.
Fig. 8. Tonic and phasic inhibition of WT and mutant rat heart
sodium channels. A and C, Current recorded during test pulses to 20 mV
in control, and during the first and one hundredth pulse of 5 Hz trains
in a cell expressing rhWT channels (A) and rhF1762A (C) channels. The protocol
was the same as that of Fig. 1 except that holding and test potentials
were 120 mV and 20 mV, respectively, to account for the more negative
voltage dependence of the heart channel. B and D, Concentration-response
curves for tonic (closed circles) and phasic (open circles) block of rhWT
(B) and rhF1762A (D) by R-mexiletine determined from pulse 1 and pulse
100 of trains as described in the legend to Fig. 1. The solid lines are
fits of a logistic equation with IC50 values for rhWT (B) of 165 µM
and 52 µM for tonic and phasic block, respectively, and for rhF1762A
(D) of 748 µM and 618 µM. The dotted lines are the fit curves
to the analogous rbWT results from Fig. 1B and from rbF1764A from Fig.
1D.
Fig. 9. Frequency-dependent inhibition of rhWT and rhF1762A mutant
sodium channels. A, Cells expressing rhWT sodium channels were stimulated
with trains of 15 depolarizations (10 ms duration) to 20 mV from a holding
potential of 120 mV at the indicated frequencies in the presence of 100
µM mexiletine. The current evoked by pulse 15 is plotted as a fraction
of the peak current evoked by pulse 1 (the tonically blocked channel) for
each stimulus frequency. B, The pulsewise development of frequency-dependent
block using 5 Hz stimulation was for WT (circles) and rhF1762A (squares)
channels. The solid lines are exponential fits to the data with rate constants
of 0.39/pulse for WT and 0.42/pulse for rhF1762A. For better comparison
of pulse-dependent development of block the rhF1762A curve was scaled vertically
to match the magnitude of block in rhWT (dotted line).
Fig. 10. Depolarization-dependent block of heart sodium chanels
by R-mexiletine. A and B, The time course of onset of R-mexiletine block
during depolarization was studied using a prepulse to 20 mV of variable
duration followed by a test pulse to the same potential. A 45 ms interval
was present between prepulse and test pulse to allow of non-drug-bound
channels to recover from fast inactivation. Peak test pulse current in
the presence of 10 mM (circles), 30 mM
(triangles), 100 mM (inverted triangles), 300
mM
(squares) and 1000 mM (diamonds) R-mexiletine
was normalized to control values obtained using the same protocol and plotted
versus prepulse duration for cells expressing rhWT (A) or rhF1762A (B)
sodium channels. Note the different range of concentrations for rhWT and
rhF1762A. C and D, Comparison of the onset of block by 300 µM R-mexiletine
for rbWT and rbF1764A (C; see Fig. 3A,B), and rhWT and rhF1762A channels
(D), respectively. Shown are fits of two exponentials to the data points.
The first and last points of the curves were set to 1 and 0, respectively,
to compare the kinetics of development of block. The mutant F1764A had
a much stronger effect on the onset of block (C), compared to the the equivalent
heart construct, rhF1762A (D).
Fig. 11. Mutant rhF1762A reduces block of depolarized sodium
channels. A, The inhibition of inactivated sodium channels was investigated
using a 1 s depolarizing prepulse to 60 mV. The membrane potential was
then returned to the holding potential of 120 mV for 10 ms, and a test
pulse to 20 mV was then applied. R-Mexiletine block of peak test pulse
current is plotted as a function of R-mexiletine concentration for rhWT
(circles) and rhF1762A (squares). The solid lines are fits of a logistic
equation to the data with IC50 values of 9.4 µM (rhWT) and 149 µM
(rhF1762A), respectively. The dotted lines represent logistic fits for
the corresponding brain channels (see Fig. 4), with IC50 values of 20.7
µM (rbWT), and 156 µM (rbF1764A). B, Shift in half inactivation
voltage, V0.5, as a function of R-mexiletine concentration in
rhWT (circles) and rhF1762A (squares). C, Shift in half inactivation voltage
in response to 300 µM R-mexiletine.
Fig. 12. Holding potential-dependence of concentration-response
curves at strongly hyperpolarized potentials to determine the limiting
affinity of resting channels at negative potentials. The indicated concentrations
of R-mexiletine were applied to rhWT (A) or rhF1762A (B) channels at holding
potentials of -100 mV (squares), -120 mV (triangles), -140 mV (inverted
triangles) and -160 mV (diamonds). The concentration-response curve for
each channel type reaches a limiting affinity at the most negative potentials.
Fig. 13. A,B, Recovery from use-dependent block of rhWT and rhF1762A
sodium channels. Use-dependent inhibition by 100 or 300 µM
mexiletine was generated by applying a 10 Hz conditioning train of 10 pulses
to 0 mV. The membrane potential was then returned to -120 mV and a test
pulse to -20 mV was applied after a recovery interval of variable duration.
The rates of recovery at -120 mV for WT (A) and mutant rbF1762A (B) were
generated by plotting the peak test pulse current normalized to its value
after 5000 ms recovery as a function of recovery interval. The data were
fitted by double-exponential functions, where the faster time constant
reflected the recovery from inactivation of unblocked channels, and the
slower, the recovery from block by mexiletine. This slower time constant
was 529 ms for rhWT, but only 168 ms for rhF1762A.
Fig. 14. Recovery from inactivation is slowed in mutant rhT1755V.
The pulse protocol was identical to that of Fig. 13. Fits of a double exponential
function to the recovery data gave a time constant for the slow component
of 784 ms. The fit to the recovery data from rhWT channels is shown for
comparison (dotted line).
index terms
sodium channels, local anesthetic, antiarrhythmic, heart, brain, channel
blocker

{kind=link}
{kind=link}