

The Mougous Lab
Defining the mechanisms and significance of interbacterial interactions in medical and environmental settings
Our Research

Antibacterial
Toxins
These are fascinating proteins that disable target cells through diverse mechanisms, providing insight into vulnerabilities of bacteria.

Epibiotic
Bacteria
These remarkable, understudied, miniscule bacteria grow on the surface of host bacteria.

Gut Bacteria Interactions
Antagonism and defense are critical for the survival of bacteria in the densely colonized mammalian GI tract.
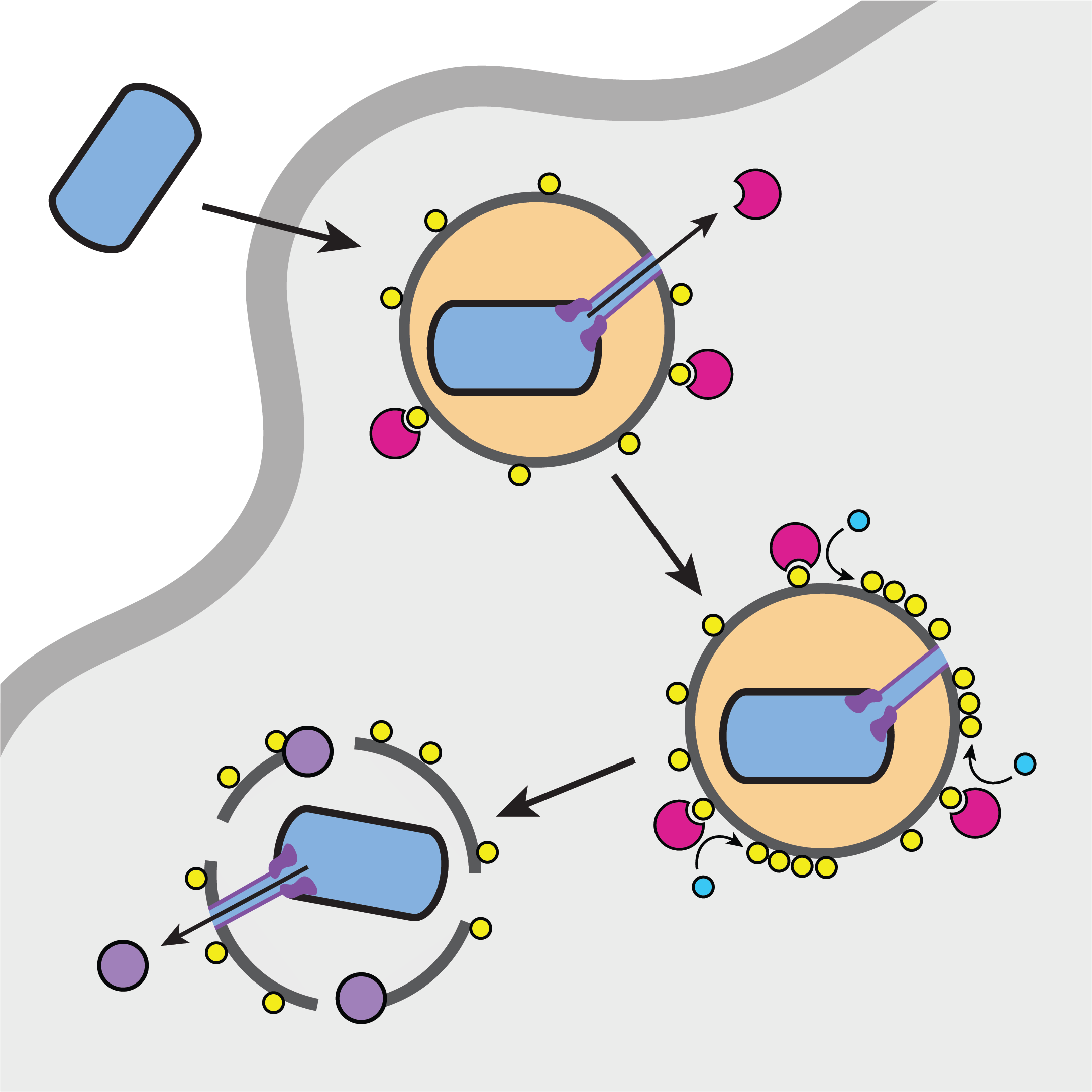
Secreted Virulence Factors
Bacteria employ toxins delivered by specialized secretions systems to overcome host defense strategies.

News
April 4, 2024 – Billy receives NSF GRFP award
Our second-year PhD student Billy Ngo has been selected for the NSF Graduate Research Fellowship. This award provides three years of funding towards Billy’s PhD. Congratulations Billy!
December 1, 2023 - Kevin successfully defends his PhD thesis
Kevin Cutler defended his thesis with a unique blend of physics and microbiology. Titled "Leveraging Machine Learning for Image Cytometry," his talk highlighted the major advancements he has made in the world of image segmentation with Omnipose. Congrats to Dr. Cutler!
November 13, 2023 - Congrats to David for successfully passing the candidacy exam
We are thrilled to announce that our MCB PhD student candidate David Brinkley has aced his qualifying exam. Well done, David!
August 18, 2023 - Farewell to our BioSTEP Summer Scholar, Marieke Trabant
Marieke had a productive summer working with Pooja on developing new peptide therapeutics, but, as summer comes to an end, we must bid her adieu. She will return to Western Washington University to finish her undergraduate degree.
June 17, 2023 - Billy Ngo and Otto Chipashvili join the lab as new graduate students
We are excited to bring on a double-dose of talent with two new graduate students from the Department of Microbiology at UW. They bring distinct expertise to the lab – Otto with a history of oral microbiome research from the Forsyth Institute, and Billy who previously studied host-microbe interactions in colon and liver diseases at UNC Chapel Hill.
June 13, 2023 - New center launched for the study of microbiomes and microbial interactions
Joseph and Kyle will be directing the new Microbial Interactions & Microbiome Center (mim_c), with the goal of advancing microbiome sciences at the University of Washington and neighboring institutions.
May 15, 2023 - Pooja Srinivas joins the lab as a new Postdoctoral Scholar
Pooja comes to us from the Dunham Lab at Emory University, where she worked on various questions pertaining to ribosome structure/function. We are excited to bring on Pooja as the newest addition to our research team.
Questions? Contact Us.
Mougous Lab/Room F510 UW Medicine at SLU 750 Republican St. Seattle, WA 98109
(206) 685-6740