I am Virginia and Prentice Bloedel Endowed Professor in the
School of Engineering and Technology at University of Washington - Tacoma, and
an Adjunct Professor in the Department of Microbiology at University of Washington - Seattle.
My research focuses on the development of optimized methods and cloud-based software tools for the analyses of big biomedical data.
My group has expertise in next generation sequencing, long read sequencing and imaging data.
My research experience includes bioinformatics, machine learning, data science, systems biology, cancer genomics, and precision medicine.
I am a computer scientist by training (Ph.D. in Computer Science from
University of Washington - Seattle
under the supervision of
Larry Ruzzo), and have extensive experience in interdisciplinary collaborative projects.
Our lab is part of the DRACC (Data Resource Administrative Coordination Center) in the NIH MorPhiC consortium.
We are primarily in charge of developing analytical pipelines to process diverse datasets generated in the NIH MorPhiC consortium.
The first marker (perspective) paper for MorPhiC is published in the February 13, 2025 release of Nature.
We are recruiting PhD, Master's, and undergraduate students interested in applying computational techniques to bioinformatics applications.
Please reach out to me via email if interested.
Presentation of Distinguished Research Award at University of Washington Tacoma Video. Oct 4, 2019.
New software will transform biological data analysis. An NIH-funded team at UW Tacoma, led by Dr. Ka Yee Yeung, has launched BioDepot-workflow-builder to move bioinformatics data analysis into the cloud.
Sept 26, 2019.
Full article.
UW Tacoma's Dr. Ka Yee Yeung-Rhee, Ph.D, is working with Dr. Pamela Becker, a UW Medicine hematologist who specializes in blood cancer research. Dr. Yeung-Rhee, a big-data expert, is looking for statistical patterns in cancer patients' genetic mutations and the drugs they respond to.
More
Madigan pairs with UW to improve medicine, by Suzanne Ovel, Northwest Guardian, Jan. 5, 2017.
Full article
Research Highlights
NIH MorPhiC consortium:
I serve as a MPI in the DRACC (Data Resource Administrative Coordination Center) of NIH-funded MorPhiC Consortium from 2022 to present.
The MorPhiC (Molecular Phenotypes of Null Alleles in Cells) program aims to develop a consistent catalog of molecular and cellular phenotypes for null alleles for every human gene.
The DRACC coordinates the data to ensure FAIRness, storage and distribution.
In particular, our team at University of Washington is primarily in charge of data processing.
Related manuscript(s): Nature 638, 351-359 (2025) Related Resources: MorPhiC web and data portal.
MorPhiC GitHub,
UWT News May 2023
Fast, portable and affordable cancer diagnostics:
In collaboration with the Fred Hutchinson Cancer Center, we develop integrated bioinformatics solution to support the analyses of long-read sequencing data.
Coupled with fast, portable and affordable sequencing technologies, we aim to deliver cancer diagnostics with a same-day turnaround.
Related manuscripts: PLOS Global Public Health 2023,
BMC Genomics 2021
Network inference from diverse genomics data:
Interactions among genes and their gene products comprise a regulatory network.
The goal of network inference is to generate testable hypotheses of gene-to-gene influences and subsequently design bench experiments to confirm network predictions.
In the November 2011 issue of PNAS, Yeung and colleagues presented a methodology to construct gene
regulatory networks from time series expression data in yeast,
integrating various types
of external biological knowledge available from public repositories.
We generated microarray data measuring time-dependent gene-expression levels in
95 genotyped yeast segregants subjected to a drug perturbation.
Our algorithm is capable of generating feedback loops and we showed that the
inferred network recovered existing and novel regulatory relationships.
In addition, we generated independent microarray data on selected deletion
mutants to prospectively test network predictions.
Related work: BMC Systems Biology 2012,
BMC Systems Biology 2014,
bioRxiv 099036
From computational discoveries to translational research:
The development of genetic predictors of clinical outcomes contributes
to risk assessment in personalized medicine.
In collaboration with Dr. Jerry Radich and Dr. Vivian Oehler at the
Fred Hutchinson Cancer Research Center, we aim to develop computational
models that can predict patient responses to therapy at diagnosis,
which allow us to tailor therapy to individual patients of
chonic myeloid leukemia (CML).
We have previously applied Bayesian Model Averaging to a gene expression
data studying the progression of CML, and identified 6 predictive genes
in
Blood 2009.
Building on this work, we developed a network-driven approach that
uses expert knowledge and predicted functional relationships to guide
our search for signature genes in the
March 2012 issue of Bioinformatics.
We showed that our gene signatures of advanced phase CML are predictive of relapse even after adjustment for known risk factors associated with transplant outcomes.
Pattern discovery and feature selection:
I have also contributed to the development and application of
pattern discovery and feature selection in computational biology,
including clustering algorithms and supervised learning methods.
 I am Virginia and Prentice Bloedel Endowed Professor in the
School of Engineering and Technology at University of Washington - Tacoma, and
an Adjunct Professor in the Department of Microbiology at University of Washington - Seattle.
My research focuses on the development of optimized methods and cloud-based software tools for the analyses of big biomedical data.
My group has expertise in next generation sequencing, long read sequencing and imaging data.
My research experience includes bioinformatics, machine learning, data science, systems biology, cancer genomics, and precision medicine.
I am Virginia and Prentice Bloedel Endowed Professor in the
School of Engineering and Technology at University of Washington - Tacoma, and
an Adjunct Professor in the Department of Microbiology at University of Washington - Seattle.
My research focuses on the development of optimized methods and cloud-based software tools for the analyses of big biomedical data.
My group has expertise in next generation sequencing, long read sequencing and imaging data.
My research experience includes bioinformatics, machine learning, data science, systems biology, cancer genomics, and precision medicine.
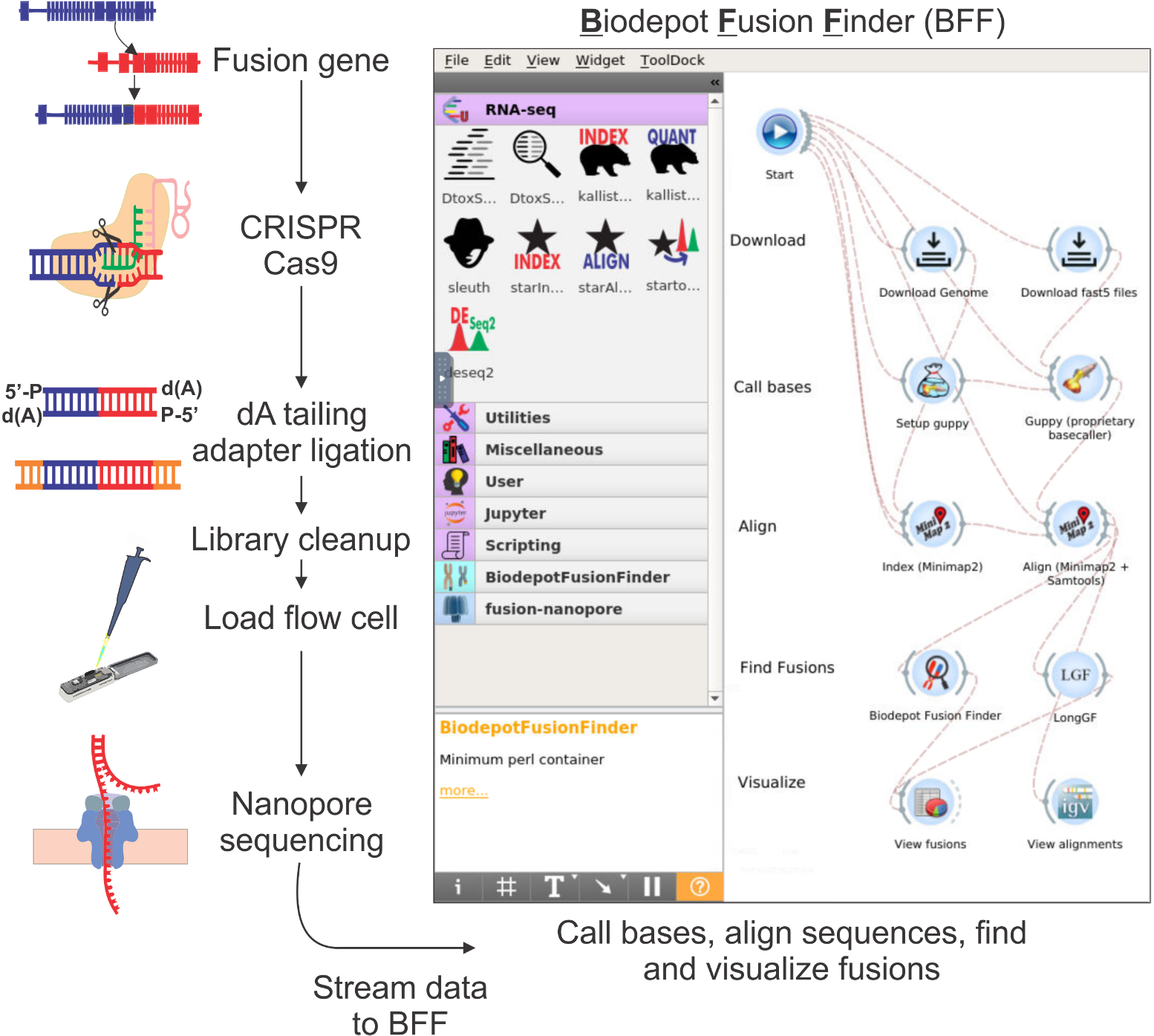 Fast, portable and affordable cancer diagnostics:
In collaboration with the Fred Hutchinson Cancer Center, we develop integrated bioinformatics solution to support the analyses of long-read sequencing data.
Coupled with fast, portable and affordable sequencing technologies, we aim to deliver cancer diagnostics with a same-day turnaround.
Fast, portable and affordable cancer diagnostics:
In collaboration with the Fred Hutchinson Cancer Center, we develop integrated bioinformatics solution to support the analyses of long-read sequencing data.
Coupled with fast, portable and affordable sequencing technologies, we aim to deliver cancer diagnostics with a same-day turnaround.
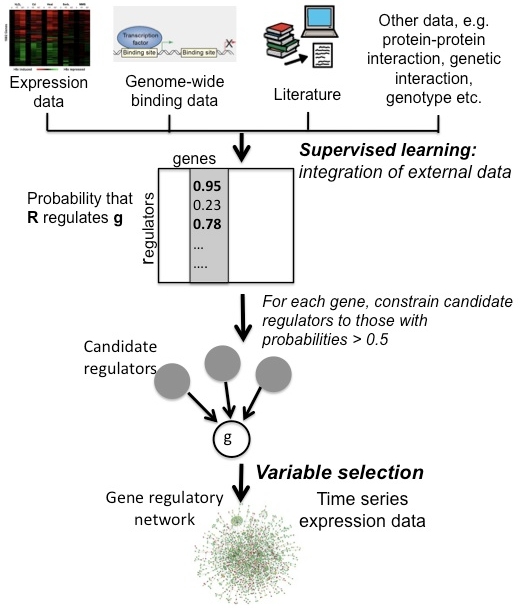 Network inference from diverse genomics data:
Interactions among genes and their gene products comprise a regulatory network.
The goal of network inference is to generate testable hypotheses of gene-to-gene influences and subsequently design bench experiments to confirm network predictions.
In the
Network inference from diverse genomics data:
Interactions among genes and their gene products comprise a regulatory network.
The goal of network inference is to generate testable hypotheses of gene-to-gene influences and subsequently design bench experiments to confirm network predictions.
In the 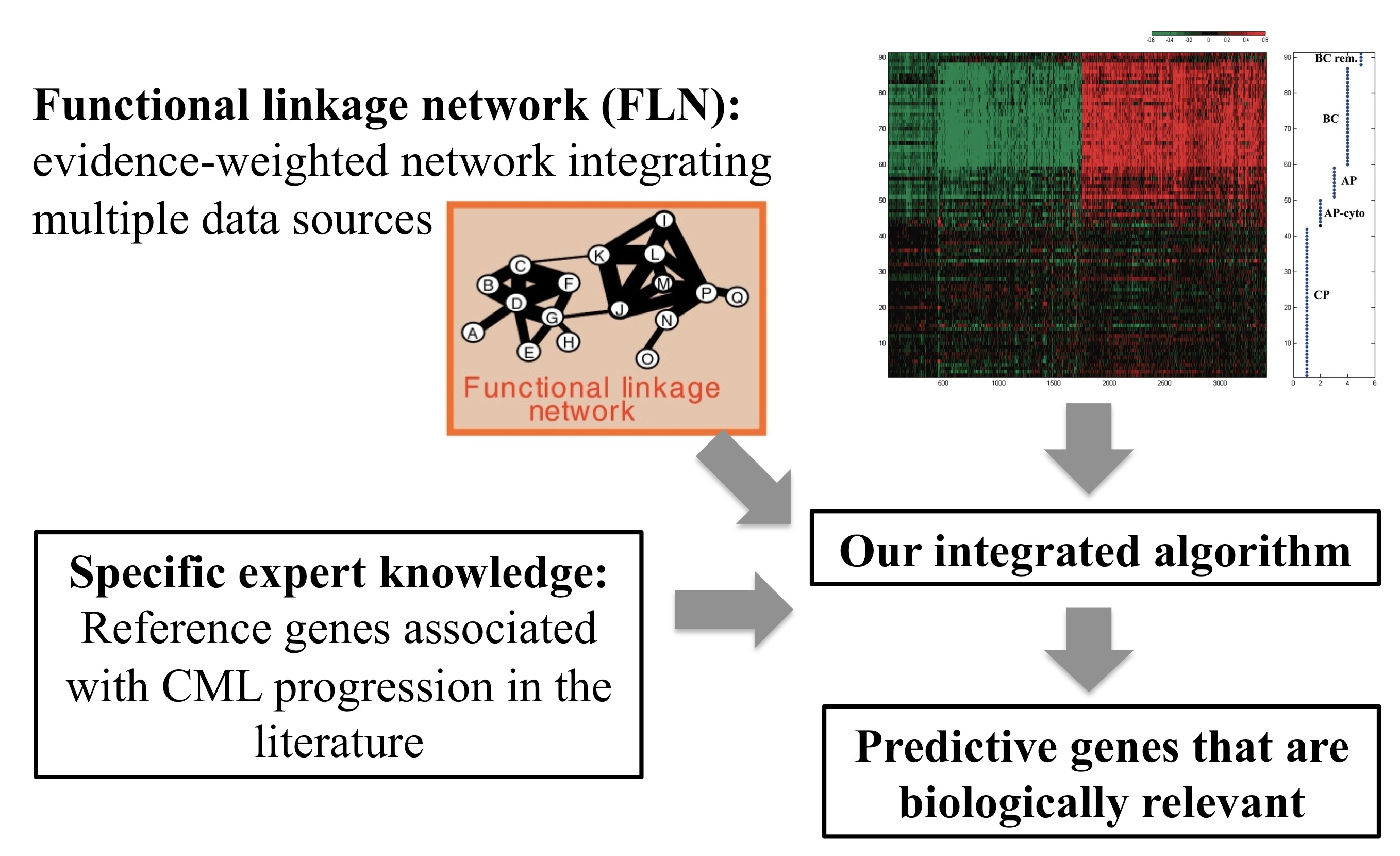 From computational discoveries to translational research:
The development of genetic predictors of clinical outcomes contributes
to risk assessment in personalized medicine.
In collaboration with Dr. Jerry Radich and Dr. Vivian Oehler at the
Fred Hutchinson Cancer Research Center, we aim to develop computational
models that can predict patient responses to therapy at diagnosis,
which allow us to tailor therapy to individual patients of
chonic myeloid leukemia (CML).
We have previously applied Bayesian Model Averaging to a gene expression
data studying the progression of CML, and identified 6 predictive genes
in
From computational discoveries to translational research:
The development of genetic predictors of clinical outcomes contributes
to risk assessment in personalized medicine.
In collaboration with Dr. Jerry Radich and Dr. Vivian Oehler at the
Fred Hutchinson Cancer Research Center, we aim to develop computational
models that can predict patient responses to therapy at diagnosis,
which allow us to tailor therapy to individual patients of
chonic myeloid leukemia (CML).
We have previously applied Bayesian Model Averaging to a gene expression
data studying the progression of CML, and identified 6 predictive genes
in