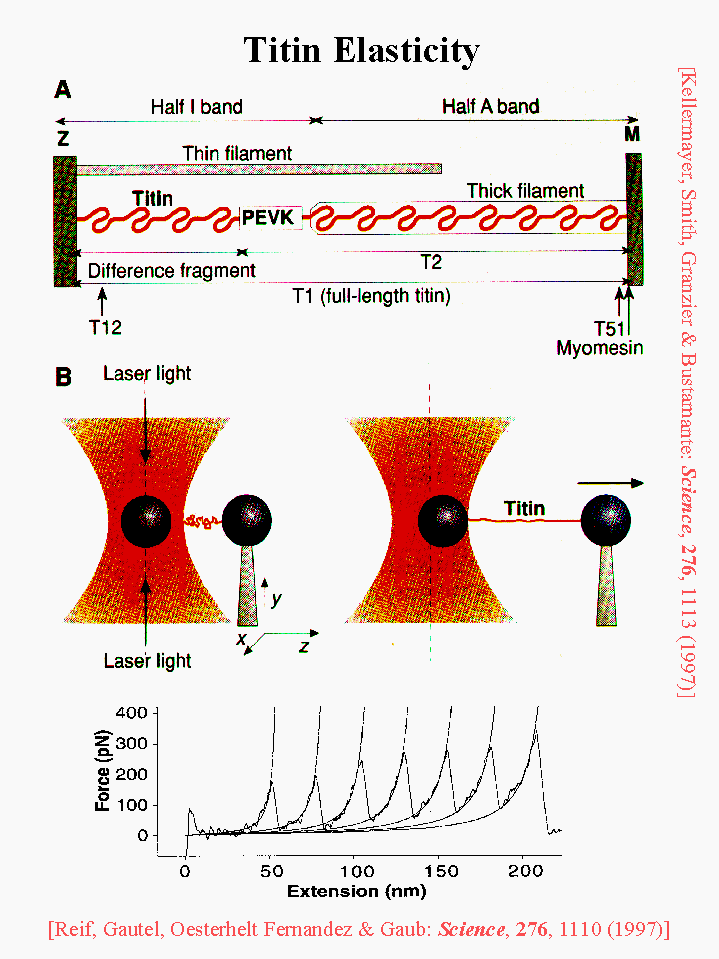
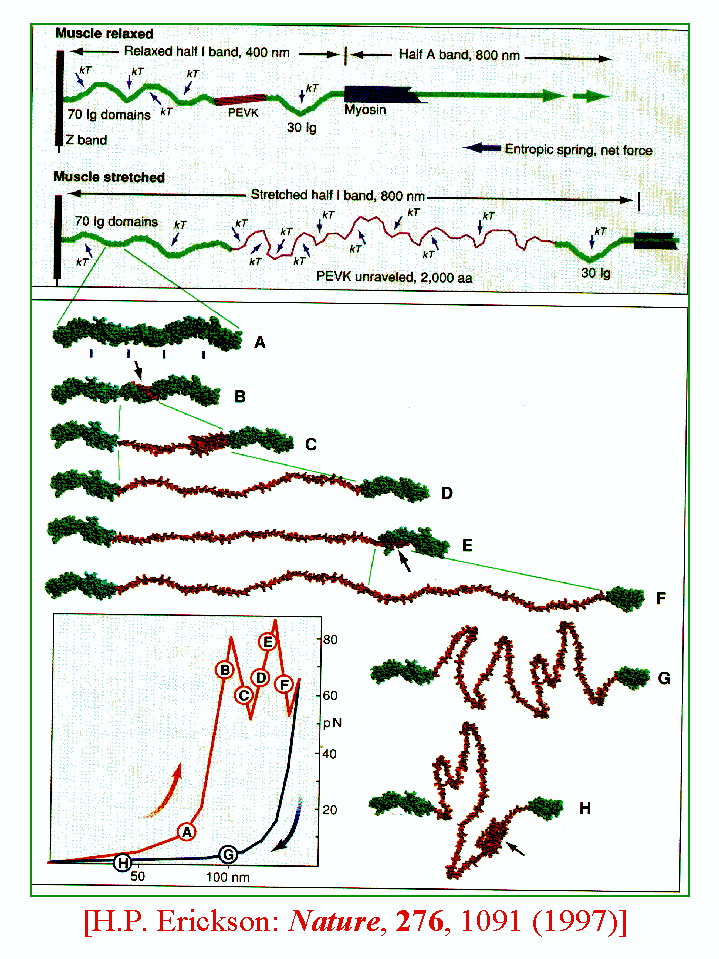
An Analysis of the
Thermodynamics of Hydrophobic Solvation Based on Scaled Particle
Theory
Adaptive Calcium Dynamics in Cardiomyocytes and Calcium-Induced
Calcium Release
An adaptive kinetic model for the calcium induced calcium release
channel (also known as the ryanodine receptor) is studied. The model
incorporates two characteristic calcium concentration dependencies
indicated by experimental observations; namely, adaptive kinetic
activation in a sub-micromolar range and equilibrium activation
in a micromolar range. Following an equilibrium analysis, the kinetics
of the model is cast in and analysed by a 3-state approximation, which
yields an analytical solution. Temporal inactivation, as suggested
by experimental measurements from skinned cardiac myocytes, naturally
emerges from the single channel kinetics. The kinetic model
of a single channel is then used to discuss the calcium
induced calcium release in a cellular compartment. We show that the
adaptive model for a single channel exhibits oscillatory calcium dynamics
{\it in situ}. We conclude that temporal inactivation without
concentration inhibition is sufficient as a negative control for rich
calcium dynamics in cells.
|
Brownian Ratchet and Protein Polymerization Against Force
The growth of filamentous proteins such as actin and microtubules
can do work against molecular or cellular barriers that resist movement;
hence it has been suggested that these filaments are partially
responsible for the shape of various biological cells, and their dynamics
are partially responsible for the extension of lamellipodia of motile
cells (Cortese et al., 1989; Felder and Elson, 1990). To understand the
physical principles behind this biochemical process, T.L. Hill has
developed an elegant thermodynamic theory, for both equilibrium and
nonequilibiurm, of polymerization against force (Hill, 1981, 1987).
Recently, a more mechanistic model termed the {\it Brownian ratchet} (BR)
has appeared (Peskin et al., 1993). A detailed mathematical analysis
has revealed the crucial role of thermal fluctuations of both the
filamental lengthening and the barrier; a true fluctuating molecular
picture emerged. The model developed by Peskin et al.
is essentially a theory for single filament; from their mathematical
formalism it is not clear how to generalize the model to account for
multiple filaments in a bundle, a situation which is biologically
more realistic. The issue of how the multiple filaments
grow as a bundle has important relevance in cell biology.
|
DNA Supercoiling and Nonlinear Elasticity
It is well known that a large linking number induces an
abrupt writhing of a circular rod with zero intrinsic curvature, i.e.,
the stress-free state of the rod is straight. We show here that for
any rod with a uniform natural curvature, no matter how small the
intrinsic curvature is, a twist will induce a continuous writhing
from the circular configuration and the abrupt writhing is only the
limiting case when the intrinsic curvature is absolutely zero. The
implication of this result on elastic models of circular DNA is
discussed.
|
Energy Balance and Purine Nucleotide Metabolism in Muscle
Purine nucleotides are among the most important metabolites
in cellular biochemistry. In addition to their well-known roles
as precursors and intermediates of various biosynthesis and major
players of cellular energetics, they are also involved in linking
the energy balance of cells to the putative functional
regulation of vasculature in cardiac and skeletal muscles. However,
the precise mechanism(s) by which these regulations are carried
out and in fact their significances {\it in vivo} remain to be a
unsolved issue in both multi-cellular biochemistry and physiology
(Schrader et al., 1998). These questions are
particularly important in skeletal muscle cells and cardiac tissues
where energy and its balance are the primary concerns of the cells.
Under a normal physiological condition, the energetics inside these
cells is balanced - the ATP synthesis and hydrolysis are well matched,
and the level of total adenine nucleotide inside the cell is constant
and dominated by ATP and ADP. The total concentration of AMP, adenosine,
IMP, and their metabolites (inosine, hypoxanthine, etc.,) are low. When
muscle cells undergo short normal exercise, the ATP-ADP dynamics are
controlled by the action of creatine kinase, serving as a short-term
buffer for cellular ATP (McFarland et al., 1994).
In the present work, we develop a parallel model to study the
energy balance in muscle and the purine metabolism. The model
consists five components (Fig. 1B): ATP synthesis by oxidative
phosphorylation, ATP hydrolysis, creatine kinase (CK), myokinase (MK),
and purine nulceotide cycle (PNC). In contrast to the cardiac
purine metabolism, this system is closed with respect to purine.
The AMP deaminase irreversibly converts AMP to IMP, then the PNC
regenerates AMP from IMP on a slower time scale. As we shall show,
this temporary exclusion of purine, in the form of IMP, from the
ATP-ADP dynamics has a similar effect as the open adenylate system
but without a permanent lost of purine (Meyer and Terjung, 1980b).
This model is a further development of the simple, three-component
model for muscle energy balance recently proposed by Kushmerick (1998).
Fig. 1B has two additional components: myokinase and purine
nucleotide cycle.
|
Protein Folding Kinetics and Thermodynamics
A simple energy landscape for protein folding (Bryngelson et al.,
1995) is described. The model emphasizes the biased diffusive nature
of protein folding (Levinthal, 1969) by utilizing the concept of
dimensional reduction (Adam and Delbr\"{u}ck, 1968) which fits the
recent observed folding intermediates in pulse-labeling hydrogen
exchange experiments (Baldwin, 1993). The model has no enthalpic
barriers between folded and unfolded protein. However an entropic
barrier evidently gives rise to a two-state global folding kinetics.
It is argued that the intermediate is in fact the transition state for
the folding kinetics. The folding pathway should be understood as a
sequence of events which selectively reduce the degrees of freedom of
the peptide chain.
|
Stochastic Mechanics and Dynamics of Single Motor Proteins
We carry out a theoretical analysis for the force generation of
a single motor protein against an elastic load. The analysis is based
on a previous work in which a simple kinetic framework for studying motor
protein movement is developed (Qian, 1997). In particular, the kinetics
of ATP hydrolysis and the stochastic movement of a motor protein along
its track have been integrated into a coherent picture. We show that the
stochastic motion (Svoboda et al., 1994; Meyh\"{o}fer and Howard, 1995)
of the single motor protein is an inhomogeneous random walk in a harmonic
potential. We further show that the dynamic force-velocity relationship
obtained from the single motor measurements is equivalent to the isotonic
velocity. The force-velocity relation is predicated to have curvature but
appears to be linear in some parameter region. The transient relaxation
time, the amplitude and correlation time of the stationary fluctuation under
isometric condition can all be obtained. When considering the maximal
force generated by the motor protein at both high and low ATP
concentrations, the model fails to provide the [ATP] independent
maximal force observed in experiments. Detailed analysis indicates
that multiple kinetic cycles, some of them being futile, must be
present in order to address the [ATP] dependence issue. Some
suggestion for experimental work is provided.
|